Basti considerare tra tutti la rilevanza che il progetto Genoma Umano ha avuto ed ha, anche come interesse per la pubblica opinione in seguito alle possibili ricadute in campo biologico, medico, patologico e bioetico. Inoltre, accanto all'archiviazione dei dati, diventa fondamentale la possibilitŗ di ricavare informazione in modo automatico dalle banche dati.
Molteplici sono gli scopi, tra cui la possibilitŗ di ricostruire le tappe evolutive delle varie specie, incluso l'uomo, e la possibilitŗ di ricavare caratteristiche per le varie biomolecole utili nella progettazione di nuove molecole e farmaci in settori diversi, dall'agro-alimentare a quello farmaceutico.
Grazie alla Bioinformatica la ricerca biologica e biotecnologia puÚ essere direzionata e mirata, con notevole riduzione nei costi attuativi. Infine sta assumendo un ruolo sempre piý centrale, sia per quanto riguarda la gestione e l'integrazione dei dati biologici (sequenze di DNA e proteine, strutture proteiche, ecc.), sia per l'elaborazione dell'informazione che molto spesso richiede lo sviluppo di procedure informatiche e algoritmi nuovi. In questa tesina verrŗ spiegata la tecnica del Mappaggio del DNA, ovvero la tecnica che ci permette di riconoscere e dividere sequenze specifiche di DNA basandosi sull'azione degli enzimi di restrizione e poi vedremo come gli Algoritmi a Forza Bruta sono di ausilio alle tecniche del mappaggio. Infatti questo tipo di algoritmi prendono in input delle sequenze di DNA che vengono analizzate tutte fino a trovare quella cercata.
Coscienti dellíimportanza del DNA nellíevoluzione della medicina, cercheremo di illustrare a persone non del campo medico, come líinformatica applicata alla biologia molecolare possiede delle potenzialitŗ che saranno fondamentali per un futuro díinnovazione.
- Nel primo capitolo verranno evidenziate le tecniche fondamentali per la scoperta di particolari sequenze del DNA e che hanno reso possibile la ricerca su di esso, che come sappiamo contiene tutto il nostro patrimonio genetico e quindi di vitale importanza per líindividuazione di nuove tecnologie che andranno a migliorare la nostra vita.
- Nel secondo capitolo ci dedicheremo a dei particolari enzimi, detti ďdi restrizioneĒ, che hanno un ruolo preponderante nelle tecniche di mappaggio del DNA. Infatti solo dopo la loro scoperta si Ť resa possibile uníinnovazione nella ricerca sulle molecole di DNA, infatti grazie alla loro azione Ť possibile riconoscere e dividere le sequenze di DNA in particolari punti detti appunto di restrizione.
- Nel terzo capitolo tratteremo la sintesi parziale di una sequenza di DNA, ovvero il processo nel quale l'informazione contenuta nel DNA dei geni strutturali viene trasformata in proteine che vanno a formare ed a far vivere una cellula. Con la sintesi parziale conosciamo poi le distanze tra i punti di restrizione calcolati nel capitolo precedente.
- Infine nel quarto capitolo affronteremo gli algoritmi che sono di ausilio alle tecniche giŗ esposte nei capitoli precedenti, cercando di mostrare in che modo líinformatica e quanto piý gli algoritmi, risultano utili alla risoluzione di problematiche riguardanti il mappaggio del DNA.Analizzeremo gli algoritmi a Forza Bruta i quali esaminano le sequenze omonime in una sequenza data di DNA.
Innanzitutto cominciamo con il capire cosíŤ il DNA. Abbreviativo di acido desossiribonucleico Ť un polimero di unitŗ piý piccole legate tra loro attraverso legami fosfo-diesterici: i nucleotidi. Abbiamo 4 diversi tipi di nucleotidi, che si differenziano per la base azotata che contengono. Infatti un nucleotide Ť costituito da uno zucchero (il deossiribosio nel DNA o il ribosio nell'RNA), un gruppo fosfato, ed una base azotata (citosina, adenina, timina, uracile, guanina). La struttura del DNA Ť stata scoperta nel 1953 da Watson & Crick: il doppio filamento Ť costituito da due filamenti anti paralleli (e quindi con direzioni opposte, 5'-3' e 3'-5'), attorno ad un asse centrale. In funzione della composizione del filamento dell'idratazione, abbiamo diverse forme. Si distinguono infatti isoforme A, B, Z dove sono diversi alcuni parametri strutturali, come il diametro della doppia elica, o la distanza tra un filamento e l'altro.. A causa di proteine dette istoniche che si associano con il DNA, il filamento si organizza in nucleosomi, e quste si strutturano in polinucleosomi. Essi poi si condensano ulteriormente in nucleofilamenti, che si associano in anse su una struttura polisaccaridica, detta scaffold, che costituisce proprio lo scheletro del cromosoma.
Nella figura seguente vediamo la struttura delle diverse componenti del DNA a partire dal cromosoma quindi la struttura a doppia elica allíinterno della quale ci sono le basi azotate.

L'autoradiografia Ť impiegata nello studio e nella localizzazione del DNA, usando come traccianti degli acidi che contengono isotopi radioattivi, soprattutto trizio, come per esempio la timidina. Negli anni Ottanta del sec. XX l'autoradiografia ha avuto un ulteriore sviluppo grazie alla scoperta di nuovi traccianti radioattivi, metodi di risoluzione delle immagini piý sensibili e, soprattutto, lettori di immagini collegati ai computer che hanno permesso di ottenere misure quantitative molto precise della radioattivitŗ di un campione. … infatti stato possibile misurare il consumo di glucosio in molti tessuti, incluso il cervello, come un indice della loro attivitŗ metabolica, rivelando quindi stati patologici. In farmacologia, l'autoradiografia Ť impiegata per studiare il legame dei farmaci ai recettori attraverso i quali esplicano la loro azione.
In biologia molecolare Ť comunemente utilizzata per evidenziare l'ibridazione di una sonda radioattiva con RNA o DNA denaturato, in vari esperimenti quali Southern blot, Northern blot, ibridazione di colonie batteriche o di placche fagiche. Un altro metodo per visualizzare strisce di DNA sul gel Ť la fluorescenza che consiste nellíincubare il gel in una soluzione contenente una sostanza colorante: líethidium che si lega al DNA che a sua volta si illumina quando il gel Ť esposto a luce ultravioletta. La fluorescenza emessa Ť letta mediante una camera CCD (charge-coupled device) ad altissima risoluzione (50 Ķm/pixel) che rivela l'emissione in fluorescenza dei campioni analizzati, offrendo all'utilizzatore - in pochi minuti - l'elettroferogramma (la serie di picchi colorati della seconda immagine) con la relativa sequenza. I sequenziatori vengono controllati attraverso specifici programmi che permettono di controllare i parametri elettroforetici e di gestire l'analisi e l'elaborazione dei dati. Questo tipo di sequenziatori automatici, in origine dotati di un unico capillare singolo, arrivano oggi ad alloggiare fino 96 capillari per l'analisi automatica e contemporanea di migliaia di campioni al giorno. I metodi di sequenziamento tradizionali richiedevano tempi molto lunghi, sia per la preparazione del campione, sia per la lettura dei risultati, oltre a impiegare sostanze radioattive per riuscire a rilevare la sequenza delle basi. Negli ultimi 10 anni si Ť assistito a una rivoluzione nel sequenziamento del DNA: i rapidi progressi nel campo della genomica e della ricerca genetica, resi possibili anche dalla stessa automazione di questa tecnica, richiedono ora macchinari sempre piý potenti e veloci, ad alta produttivitŗ. Al giorno d'oggi, il mercato offre un'ampia scelta di sequenziatori automatici con relativi software di analisi dei dati e, di fatto, il sequenziamento del DNA viene effettuato per lo piý con questi apparecchi dotati di sistemi ad alta sensibilitŗ. Si riescono infatti a produrre ottimi risultati anche a partire da bassissime quantitŗ di DNA.
Nella figura successiva vediamo le bande che rappresentano segmenti di DNA progressivamente piý lunghi andando dal basso verso l'alto; le varie colonne si riferiscono alle diverse reazioni di rottura chimica secondo Maxam e Gilbert. La prima colonna contiene i frammenti che terminano con T, la seconda con C, la terza con A (e talvolta con C), la quarta con A (e talvolta con G), la quinta con G. La separazione Ť stata ottenuta per elettroforesi su gel di poliacrilammide. La sequenza Ť indicata a destra: in basso, accanto alle triplette codificanti sono indicati gli amminoacidi corrispondenti.

Nella figura seguente appunto vediamo come si isola il filamento di DNA in un campo elettrico tra le maglie di agarosio.

CAPITOLO 2 - ENZIMI DI RESTRIZIONE
In questo capitolo parleremo degli enzimi di restrizione. Eí stata fondamentale la scoperta degli enzimi di restrizione nella ricerca, soprattutto quella riguardante il DNA, infatti questi enzimi sono capaci di riconoscere specifiche sequenze di DNA e permettono di tagliare il filamento in posizioni ben precise. Si capisce quindi líimportanza della scoperta del primo enzima di restrizione che fu fatta da Hamilton Smith(1) nel 1970 dal batterio Haemophilus influenzae.Lo scoprž casualmente mentre studiava come líenzima Haemophilus influenzae raccoglie la sequenza del DNA dal virus P22. Furono riconosciute le due sequenze seguenti:
ēGTGCAC
ēGTTAAC
La sua scoperta avvenne tramite uníosservazione: quando in un ceppo del batterio Escherichia coli si introduceva del DNA proveniente da un ceppo diverso, questo DNA veniva letteralmente tagliato a pezzetti.
Fu postulato che vi fossero degli enzimi prodotti dai batteri responsabili di questa attivitŗ.
In seguito fu dimostrato che lo stesso accadeva quando i batteri venivano infettati da un virus: il DNA del virus veniva immancabilmente ridotto in frammenti inattivi, cioŤ la crescita del virus si disse ristretta in un determinato ceppo di batteri.
Di qui il nome di enzimi "di restrizione". Gli enzimi di restrizione sono ritenuti essere una sorta di sistema immunitario primitivo dei batteri.
Come fu successivamente scoperto essi attaccano solamente il DNA esterno, ad esempio di un virus, perchť il DNA dei batteri presenta delle modificazioni chimiche, per la precisione líaggiunta del gruppo metile (CH3), che lo rendono inattaccabile dai propri enzimi. Non passÚ molto tempo e il primo di questi enzimi venne scoperto e purificato in batteri della specie Haemophilus influenzae e quindi battezzato HindII.
Attualmente il loro numero ha superato abbondantemente il migliaio e sono venduti da industrie biotecnologiche che devono il loro successo al grande uso che si fa di essi nella ricerca.
Líenzima di restrizione Ť un particolare tipo di enzima che Ť in grado di riconoscere specifiche sequenze di basi lungo il filamento di DNA, e di "tagliare" esattamente in corrispondenza di queste sequenze.
La caratteristica che rende cosž preziosi gli enzimi per lo studio del DNA Ť che ognuno di essi riconosce, lega e taglia il DNA in una sequenza ben precisa.
Ad esempio, EcoRI (isolato da Escherichia coli RY13) riconosce la sequenza GAATTC, mentreBamHI (isolato da Bacillus amyloliquefaciens H) la sequenza GGATCC.
Sebbene le sequenze riconosciute siano alle volte simili, ogni enzima Ť in grado di discriminarle con estrema precisione. In questa maniera Ť possibile tagliare líintero genoma umano in frammenti piý maneggiabili, detti frammenti di restrizione; o, conoscendone la sequenza, ritagliarne fuori un certo gene; o ancora, modificare un gene accorciandolo; operazioni queste che preludono a qualunque studio di biologia molecolare.
Mediante líazione di un enzima detto DNA ligasi, i frammenti possono essere poi saldati ad un altro frammento di DNA di qualunque natura, purchŤ trattato con lo stesso enzima di restrizione (ciÚ da origine a estremitŗ complementari, che possono poi unirsi).
La possibilitŗ di costruire in vitro molecole ibride di DNA Ť stata determinata dalla scoperta di enzimi, le endonucleasi di restrizione, che tagliano la molecola di DNA a livello di siti specifici, dando cosž origine a frammenti particolari.
La maggior parte dei batteri produce uno o piý enzimi di restrizione, probabilmente perchť essi offrono il vantaggio selettivo di proteggere in qualche misura la cellula dallíinfezione da parte dei fagi che contengono DNA a doppia elica.
Finora sono state caratterizzate oltre 2000 endonucleasi di restrizione differenti, che vengono identificate con una sigla di tre lettere: la prima, maiuscola, Ť la lettera iniziale del nome del Genere, e le altre due, minuscole, sono le prime due lettere della stessa specie batterica. Ad esempio, EcoR1 e HindIII sono endonucleasi di restrizione presenti rispettivamente in particolari ceppi di E. coli e di Haemophilus influenzae.
Punto di riconoscimento degli enzimi di restrizione
Come giŗ detto nel paragrafo precedente, ogni enzima di restrizione taglia il DNA in punti della sequenza ben precisi. Nella tabella che segue ne appaiono alcuni e per ogni riga cíŤ il nome dellíenzima, il microorganismo dal quale proviene e il punto di riconoscimento dellíenzima, cioŤ la sequenza di DNA che ciascuno di questi enzimi, dopo aver riconosciuto, Ť in grado di legare e tagliare.
Uso degli enzimi di restrizione
Gli enzimi di restrizione sono importanti soprattutto, come abbiamo giŗ visto, nella tecnologia della divisione del DNA, poichť sono in grado di individuare e isolare precise sequenze di DNA. Líimportanza di questi enzimi Ť fondamentale anche per la clonazione, infatti questa tecnica anche se ancora non liberalizzata in tutti i Paesi, rappresenta il futuro della medicina con orizzonti sconfinati.Non Ť di secondaria importanza líessenziale ruolo degli enzimi di restrizione nella costruzione della biblioteca del genoma. Infine come vedremo successivamente questi enzimi saranno indispensabili nel mappaggio del DNA, combinati con le tecniche informatiche, poichť, tagliando in punti ben precisi una sequenza di DNA, saranno alla base degli algoritmi che lavorano sulla distanza tra questi punti (maggiori dettagli in seguito). Come detto poco fa ogni enzima ha un suo punto di riconoscimento, ovvero lavorando con gli enzimi di restrizione abbiamo i tagli della sequenza di DNA in punti ben precisi, una mappa di restrizione mostra la posizione di questi punti in una sequenza di DNA.
Ai primordi della biologia molecolare, le sequenze del DNA erano spesso sconosciute, infatti i biologi avevano il problema di costruire mappe di restrizione senza conoscere le sequenze del DNA. Dal momento che tali enzimi di restrizione sono molto specifici nelle loro caratteristiche e modalitŗ di taglio, questo tipo di mappa puÚ essere predetta sulla base della sequenza di un frammento noto di DNA.
Sintesi di restrizione completa
Vediamo ora come, tagliando la sequenza del DNA in ogni punto di restrizione, si creano molti frammenti di restrizione (sottosequenze). Infatti in figura vediamo rappresentato un esempio di un filamento di DNA dove sono indicati i vari punti di restrizione(Restriction Sites) dove l'enzima taglia la sequenza in sottosequenze.
Sintesi di restrizione completa: molteplici soluzioni
Come abbiamo visto dalla figura precedente, i punti di restrizione del filamento di DNA si trovavano in posizioni precise.
Supponiamo che gli enzimi di restrizione abbiano diviso la nostra sequenza di DNA in quattro frammenti di restrizione(sottosequenze di DNA) , rispettivamente di lunghezza 9, 3, 5, 5. Consideriamo ora l'ordinamento di questi frammenti con due esempi:

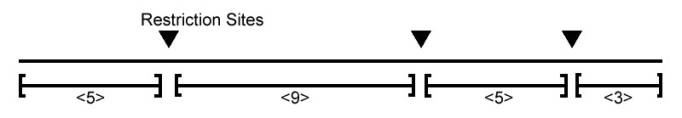
Misuramento della lunghezza dei frammenti di restrizione
Gli enzimi di restrizione interrompono la sequenza del DNA in frammenti di restrizione(sottosequenze).L'elettroforesi su gel Ť un processo per la separazione della sequenza del DNA di agarosio. L'introduzione della PFGE e le successive varianti introdotte sulla tecnica di base permettono oggi la separazione di frammenti di DNA della lunghezza di 2x103 kb. CiÚ consente la separazione in elettroforesi di cromosomi interi.
Il metodo consiste in una elettroforesi su gel di agarosio nella quale due campi elettrici con differenti angolazioni vengono applicati alternativamente per periodi di tempo definiti (ad esempŪo 60s). L'azione del primo campo elettrico causa uno stiramento lungo il piano orizzontale delle molecole avvolte e il loro movimento all'interno del gel. L'interruzione di questo campo e l'applicazione del secondo campo elettrico fa sž che le molecole si muovano nella nuova direzione. Dal momento che per una molecola a catena lunga lineare esiste una relazione tra il cambiamento conformazionale indotto da un campo elettrico e la lunghezza della molecola stessa, le molecole piý piccole si riallineeranno piý velocemente nel nuovo campo elettrico e, quindi, continueranno a muoversi attraverso il gel. Molecole piý grosse, viceversa, impiegheranno piý tempo per allinearsi. In questo modo, variando continuamente la direzione del campo, si separano le molecole piý piccole da quelle piý grandi.
PuÚ separare frammenti di DNA di diversa lunghezza in 1 nucleotide per frammento fino a frammmenti di 500 nucleotidi.
CAPITOLO 3 - PROBLEMA DI SINTESI PARZIALE
Con sintesi intendiamo una fase del processo di espressione genica, ovvero il processo nel quale l'informazione contenuta nel DNA dei geni strutturali viene trasformata in proteine che vanno a formare ed a far vivere una cellula.Nella sintesi proteica un filamento di Rna Messaggero complementare ad una data regione del DNA, Ť usato come stampo per la produzione di una specifica proteina. La relazione tra triplette di basi dell'RNA e gli amminoacidi delle proteine Ť ciÚ che chiamiamo codice genetico.
Analizziamo in questo capitolo il problema della sintesi parziale. L'esempio di DNA visto in precedenza, Ť esposto agli enzimi di restrizione soltanto per un tempo limitato per preservarlo dall'essere spezzato in tutti i suoi punti di restrizione.
Questo esperimento che stiamo per effettuare crea una collezione di tutti i possibili frammenti di restrizione tra due tagli non necessariamente consecutivi
Questa collezione di pezzi di frammenti Ť usata per determinare la posizione dei punti di restrizione nella sequenza del DNA.
Sintesi parziale: un esempio
Per la stessa sequenza di DNA, come nell'esempio precedente,considerati i seguenti punti di restrizione(Restriction Sites), vogliamo ottenere i seguenti frammenti di restrizione:
E'possibile conoscere questa molteplicitŗ di frammenti, ad esempio si puÚ determinare il numero di frammenti di restrizioni della stessa lunghezza dalla maggiore intensitŗ di fluorescenza che emette un frammento doppio rispetto a un frammento singolo.
Elementi della Sintesi parziale
Ora definiamo alcuni termini usati nel problema della sintesi parziale: Indicheremo con X la posizione di n interi rappresentanti la posizione di tutti i tagli nella mappa di restrizione, inclusi quelli di inizio e fine. Con n il numero totale di tagli e infine con DX la collezione di interi rappresentanti la lunghezza di ciascuno dei frammenti prodotti da una sintesi parzialeEsempio di sintesi parziale
Il problema della mappa di restrizione puÚ essere formulato in termini di scoperta delle posizioni dei punti solo quando conosciamo le distanze tra questi punti.Consideriamo l'insieme di elementi {2, 2, 2, 3, 3, 4, 5 }, esso Ť un multiset con elementi duplicati 2 e 3.
Se X={x1,x2,x3......,xn} Ť un insieme di n punti in ordine crescente, allora DX denota il multiset di tutte le [n(n-1)]/2 distanze tra i punti in X:
DX={xj - xi : 1<= i < j <= n}
Per esempio:
se X={0, 2, 4, 7, 10} allora la distanze tra questi punti risultano
DX={2, 2, 3, 3, 4, 5, 6, 7, 8, 10}
Nella seguente tabella riportiamo la rappresentazione della collezione di frammenti trattata in precedenza, quindi su entrambi i lati riportiamo X che Ť la posizione di tutti i punti di restrizione della nostra sequenza e nella tabella inseriamo i valori stessi della collezione.

Gli elementi (i, j) nella tabella corrispondono a |xj - xi | con 1 <= i < =j <= n.
Problema di sintesi parziale: formulazione
Adesso formuliamo il problema di sintesi parziale in questo modo: date tutte le distanze tra le coppie di punti, su una linea di DNA, ricostruire le posizioni di ciascun punto. Quindi avremo in input la collezione di distanze tra le coppie di distanze L, contenenti interi ed in output una collezione X, di n interi, tali che DX= L. Non Ť sempre possibile ricostruire in modo univoco una collezione X basata solo sull'intervallo DX.Infatti prendiamo come esempio la collezione:
A = {0, 2, 5}
e
(A 10) = {10, 12, 15}
Dove (A 10) Ť definita da {a + 10: a appartenente ad A}
queste producono entrambe {2, 3, 5} come collezione di sintesi parziale.
In generale le collezioni A e B sono dette omonime se risulta DA=DB. Consideriamo ora A={6, 7, 9} e B={-6, 2, 6} avremo A+B={0, 1, 3, 8, 9, 11, 12, 13, 15} mentre A-B={0, 1, 3, 8, 9, 11, 12, 13, 15} . Molti biologi oltre che basarsi sul problema di cercare la collezione X tale che risulti DX=L sono spesso interessati alle colezioni omonime.
Capiamo quindi l'importanza del problema di non unicitŗ nella ricostruzione di diverse collezioni. In questo capitolo affrontiamo il problema degli algoritmi a forza bruta e la loro possibilitŗ di impiego nei problemi riguardanti le sequenze di DNA visto fino ad ora. L'algoritmo a forza bruta Ť un algoritmo di ricerca che esamina tutte le possibili soluzioni fino a trovare quella valida.
L'approccio Forza Bruta si basa sull'analisi di tutte le permutazioni degli elementi di una lista, finchŤ non Ť trovata quella scelta. In genere questo algoritmo Ť raramente efficiente, di solito Ť impraticabile poichť su grandi sequenze richiede tempi di esecuzione troppo lunghi. L'algoritmo prende in input la lista L di n2 interi e restituisce la sequenza X di n interi in modo che si abbia DX=L.
Forza Bruta e sintesi parziale:
Ora definiamo i passi per il problema visto prima della sintesi parziale:1. Trovare il frammento di restrizione di lunghezza massima M.
2. Per ogni possibile soluzione stimare il corrispondente DX, ovvero la distanza tra ogni punto della sequenza e M.
3. Se DX Ť uguale al Digest parziale L (maggiori dettagli in seguito) allora X Ť la mappa di restrizione corretta.
Algoritmo a ForzaBruta
Definiti i passi per la risoluzione della sintesi parziale ora vediamo l'algoritmo che implementa la sequenza di istruzioni definite per trovare la mappa di restrizione:
1. BruteForcePDP(L, n):
2. M<--elemento massimo in L
3. for ogni sequenza di n-2 interi 0<x2...<xn-1<M
4. X<--{0,x2,...,xn-1,M}
5. Forma DX da X
6. if DX = L
7. return X
8. output "no soluzione"
Il problema ť selezionare n-2 interi, arbitrariamente, dall'intervallo da 0 a M. Osservando che tutti i punti in
X corrispondono alla stessa distanza in DX possiamo selezionare elementi distinti da L selezionando un numero minore
di elementi nell'intervalo da 0 a M.
Efficienza di ForzaBruta
Sfortunatamente la velocitŗ di questo algoritmo Ť O(M n-2), poichť esso deve esaminare tutte le possibili sequenze di posizione.Un modo per migliorare l'algoritmo Ť limitare i valori di xi a quelli che troviamo in L. Su questo principio si basa l'algoritmo che segue.
Altro Algoritmo a Forza Bruta
1. AnotherBruteForcePDP(L, n)
2. M<--maximum element in L
3. for ogni sequenza di n-2 interi 0<x2<...<xn-1<M da L
4. X<--{ 0,x2,Ö,xn-1,M }
5. Forma DX da X
6. if DX = L
7. return X
8. output "no soluzione"
Efficienza di Altro Forza Bruta
E' sicuramente piý efficiente del primo, ma risulta ancora lento. Infatti vengono esaminate meno sequenze ma il tempo di esecuzione Ť ancora esponenziale, O(n2n-4).
Il primo algoritmo impiega molto tempo , per sempio, su un input di L={2, 998, 1000}, ma il secondo algoritmo impiega molto meno tempo. In questo caso n=3 e M=1000.
Algoritmo Sintesi parziale
Definiamo D(y,X) come tutte le distanze tra il punto y e tutti i punti in X.
D(y,X)= {|y-x1|,|y-x2|,|y-xn|} con X={x1, x2,Ö, xn}
Digestparziale(L)
1. PartialDigest(L):
2. larghezza<--elemento massimo in L
3. CANCELLA (larghezza, L )
4. X<--{0, larghezza}
5. METTI (L,X)
1. METTI(L, X)
2. if L Ť vuoto
3. output X
4. return
5. y<--massimo elemento in L
6. Cancella(y,L)
7. if D(y, X ) Ť contenuto in L
8. Aggiungi y a X e rimuovi la lunghezza D(y, X) da L
9. METTI(L,X )
10. Rimuovi y da X e aggiungi la lunghezza D(y, X) a L
11. if D(larghezza-y, X ) Ť contenuto in L
12. aggiungi larghezza-y a X e rimuovi la
lunghezza D(larghezza-y, X) da L
13. METTI(L,X )
14. Rimuovi larghezza-y da X e aggiungi la
lunghezza D(larghezza-y, X ) a L
15. return
Data una sequenza L
L={2, 2, 3, 3, 4, 5, 6, 7, 8, 10}
X={0}
La dimensione di L Ť (n(n-1))/2=10, dove n Ť il numero di punti nella soluzione.
In questo caso n dev'essere 5, e ci riferiremo alle posizioni in X come x1, x2, x3, x4, e x5, da sinistra a destra sulla sequenza. Vediamo che 10 Ť la piý grande distanza in L, x5 deve essere nella posizione 10, cosž rimuovi (x5-x1)=10 da L ed inseriscilo in X.
Avremo:
L={2, 2, 3, 4, 5, 6, 7, 8}
X={0, 10}

Poichť i due casi sono simmetrici possiamo assumere x2=2.
Rimuoviamo quindi da L (x5-x2)=8 e (x1-x2)=2 e otteniamo:
L={2, 3, 3, 4, 5, 6, 7}
X={0, 2, 10}

Se x3=3, allora (x3-x2)=1 deve essere contenuto in L, ma non Ť cosž allora consideriamo x4=7.
D(y, X)=(3, 5, 7) Ť una sottosequenza di L. Rimuoviamo (x5-x4)=3, (x4-x2)=5 e (x4-x1)=7 e inseriamo 7 in X:
L={2, 3, 4, 6}
X={0, 2, 7, 10}

Anche in questo caso facciamo due scelte: x3=4 oppure x3=6 (ovvero 10-6). La distanza (x4-x3)=1 che non Ť il L, quindi abbiamo un'unica scelta x3=4.
Sfortunatamente D(y, X)={6, 4, 1, 4} non Ť una sottosequenza di L.
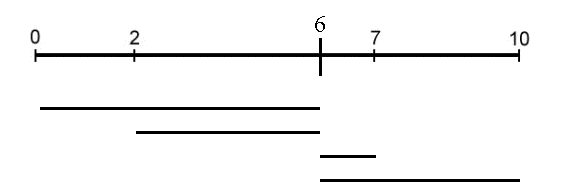
D (y, X) = {4, 2, 3 ,6}, che Ť una sottosequenza di L.
Rimuoviamo {4, 2, 3 ,6} da L e aggiungiamo 4 a X
X={0, 2, 4, 7, 10}

Analisi dell'algoritmo Digest Parziale
Per analizzare il tempo di esecuzione dell'algoritmo Metti prendiamo in considerazione le due chiamate ricorsive a 'Metti'.L'algoritmo elencherŗ tutte le sequenze X per le quali risulta DX=L. Ad ogni passo esamina due alternative, sinistra e destra.
Esso risulta essere molto veloce se consideriamo solo una delle due alternative per ogni passo.
Per molti anni non si Ť capito se il tempo di esecuzione del Digest Parziale fosse polinomiale nel caso peggiore.
Occorre tener presente che a volte entrambe le alternative sono valide.
Se entrambe, sinistra e destra, sono valide e se continuano ad esserlo nei passi successsivi, il tempo di esecuzione dell'algoritmo comincia a crescere come 2k, dove k Ť il numero di ogni passo 'ambiguo'.
Dati n punti,indichiamo con T(n) il massimo tempo necessario per trovare la soluzione.
Se si ha una sola alternativa valida ad ogni passo, l'algoritmo Digest Parziale riduce il problema chiamando se stesso ricorsivamente, cosž si ha:
T(n)=T(n-1)+O(n)
dove O(n) Ť il tempo impiegato per modificare gli insiemi X e L. Altrimenti, se si hanno due alternative valide, allora:
T(n)=2T(n-1)+O(n).
Anche se le due espressioni possono, a prima vista, sembrare simili, esse lo sono solo nella forma, infatti esprimono differenti tempi di esecuzione. La prima Ť quadratica, l'altra esponenziale.
Fino al 2002 non si conosceva un algoritmo polinomiale per il problema del Digest Parziale. Solo in quell'anno, Maurice Nivat e i suoi colleghi presentarono un algoritmo polinomiale.
Concludiamo il discorso sul Mappaggio del Dna e gli Algoritmi a Forza Bruta coscienti dell'importanza e dell'innovazione che l'informatica applicata alla biologia avrŗ un ruolo preponderante nel futuro della ricerca. ACRILAMMIDE:
composto sintetico impiegato in diversi utilizzi industriali. Ma si puÚ trovare anche negli imballaggi, nei cosmetici e nei prodotti per l'igiene personale; inoltre, si sviluppa nel fumo del tabacco.
AGAROSIO:
polisaccaride utilizzato per la produzione di gel elettroforetici;
BamHI:
Enzima di restrizione che riconosce la sequenza di coppie di basi e taglia la doppia elica in modo da produrre estremitŗ frastagliate, con basi libere alle due estremitŗ.
BASE AZOTATA:
Una delle molecole che compongono il DNA o l'RNA; sono adenina (A), guanina (G), citosina (C), timina (T) o uracile (U).
DNA:
Acido desossiribonucleico. Molecola molto lunga a due filamenti, formata dalle ripetizioni di sotto-unitŗ chiamate nucleotidi.
DNA LIGASI:
un enzima in grado di ricucire insieme i filamenti di DNA che sono stati rotti. doppia elica GAATTC.
EcoRI:
enzima di restrizione che riconosce una sequenza specifica nel DNA.
ENZIMA DI RESTRIZIONE:
Enzima capace di riconoscere un sito particolare del DNA, di fissarvisi e di realizzare un taglio dei due filamenti del DNA. Le estremitŗ del taglio possono riappaiarsi.
ENZIMA:
Proteina avente al funzione di catalizzatore. Accelera la reazione chimica per cui Ť specializzata e si ritrova intatta dopo che quest'ultima Ť stata completata.
ESCHERICHIA COLI:
sono batteri a forma di bastoncino diritto che vivono nell'ambiente intestinale dell'uomo e degli animali; sono sensibili a molti disinfettanti chimici e fisici e vengono distrutti con la pastorizzazione. L'isolamento di questi ceppi viene eseguita su terreni differenziali con successiva qualificazione attraverso test metabolici e sierologici.
GLUCOSIO:
monosaccaride molto diffuso in natura, in forma semplice oppure in molecole complesse costituite da molte unitŗ concatenate (come gli amidi, il glicogeno, la cellulosa).
GRUPPO METILE (CH3):
Questo Ť un sistema restrizione-modificazione e il suo principale scopo Ť la difesa dell'organismo.
HAEMOPHILUS INFLUENZAE:
Ť un batterio che causa una serie di malattie cosiddette invasive.
ISOTOPI RADIOATTIVI:
utilizzati per rintracciare ciÚ che accade a sostanze diverse nei processi. Mescolati con atomi non radioattivi della sostanza in esame fungono da traccianti e permettono di studiare meccanismi di reazione , di distribuzione e di assorbimento.
MRNA (RNA MESSAGGERO) :
Prodotto di trascrizione di un gene che trasporta dal DNA al citoplasma l'informazione che codifica la sequenza di una particolare catena polipeptidica. Ogni differente catena polipeptidica che la cellula sintetizza richiede la presenza di un tipo corrispondente di mRNA. Negli eucarioti, l'mRNA in genere differisce dal trascritto iniziale, a causa della eliminazione di determinate sequenze non codificanti (splicing). L' mRNA maturo, a livello ribosomiale, serve quindi da matrice per il processo di traduzione, in cui viene sintetizzata la catena polipeptidica.
NORTHERN BLOT:
tecnica utilizzata per misurare quantitativamente (quanto viene espresso) e valutare la dimensione di un RNA messaggero di un gene.
NUCLEOTIDE:
Mattone chimico elementare del DNA costituito dall'assemblaggio di una molecola di zucchero (desossiribosio), di una molecola di acido fosforico e di base azotata. Nel RNA lo zucchero Ť il ribosio.
PFGE :
tecnica che permette la separazione di frammenti di DNA della lunghezza di 2kb.
POLIACRILAMMIDE:
sostanza che si dissolve nell'acqua e viene utilizzato a livello industriale. Questi gel vengono usati per realizzare lenti a contatto morbide. L'acqua assorbita le rende morbide.
POLIMERI:
(dal greco molte parti) sono macromolecole, ovvero molecole dall'elevato peso molecolare, costituite da un gran numero di piccole molecole (i monomeri) tra loro uguali o simili unite a catena mediante la ripetizione dello stesso tipo di legame.
POLIMERIZZAZZIONE:
reazione chimica che trasforma le molecole in prodotti di reazione (cioŤ altre molecole) aventi struttura e proprietŗ differenti.
PRIMER:
corte sequenze di DNA a singola catena (circa 20-30 elementi), che vengono impiegate per duplicare un determinato tratto di DNA: prima si separano le due catene del DNA. Poi i primer si legano a singole catene di DNA.
RNA:
Acido rinucleico (Contiene ribosio). Coinvolto nella sintesi delle proteine, come intermediario del DNA, l'RNA rappresenta il materiale depositario dell'informazione genetica in alcuni virus.
SINTESI:
Preparazione di una sostanza complessa a partire dai suoi componenti.
SOUTHERN BLOT:
Tecnica utilizzata per il trasferimento di DNA su membrane su cui frammenti dello stesso DNA possono essere riconosciuti tramite l'utilizzo di sonde marcate.
TIMIDINA:
deossinucleoside costituito da timina e deossiribosio.
TRIZIO:
isotopo radioattivo dell'idrogeno.
UREA:
sostanza di scarto prodotta dal fegato ed eliminata attraverso le urine. In chimica serve per distruggere le proteine.
An Introduction to Bioinformatics Alghoritms Neil C.Jones and Pavel A.Pevzner
www.bioalgorithms.info/ presentations/Ch04_DNA_mapping.pdf
Ecco alcuni link dove Ť possibile approfondire gli argomenti trattati:
Introduction to bioinformatics - Sito del libro di testo del corso
Approfondimento sulle mappe di restrizione