
1) Estrazione di tutte le possibili parole ( words ) di w lettere (w č uguale a 12 per le query sui nucleotidi, uguale a 3 invece per le query su proteine ) dalla sequenza presa in considerazione.
Il numero totale di words presenti in una sequenza da sottoporre a confronto, risulta essere:
Le matrici di sostituzione assegnano un punteggio positivo per ogni identità o per una sostituzione con aminoacidi dello stesso tipo (idrofobici con idrofobici, carichi positivamente con carichi positivamente ecc...) e negativo per una sostituzione con aminoacidi fra loro diversi (es. aminoacido basico con aminoacido acido ecc..).
La figura mostra i passaggi 1 e 2 di BLAST
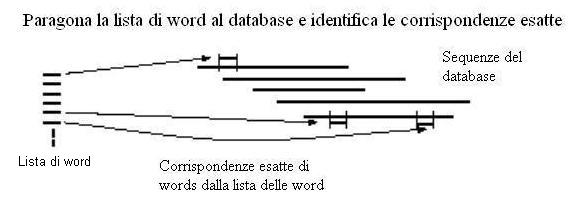
3) Ogni sequenza della banca dati viene analizzata alla ricerca di corrispondenze esatte con almeno una word della lista compilata al passo 2.
la figura mostra il passaggio 3 di BLAST
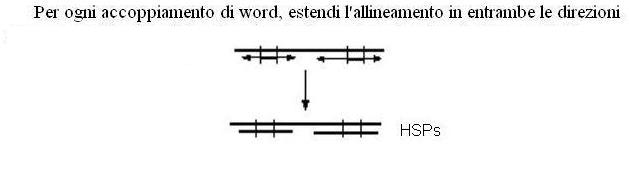
4) Estensione della zona di appaiamento. In questa fase si prendono in considerazione, uno per volta, i caratteri immediatamente adiacenti ( sia a destra che a sinistra ) alla word. Per ogni carattere aggiunto alla word si ricalcola il punteggio con la sequenza del database, e fino a quando il punteggio di similarità aumenta si aggiungeranno i caratteri adiacenti la word ( le zone di appaiamento così trovate prendono il nome di HSP – high scoring segment pair).
La figura mostra il passaggio 4 di BLAST
Per maggiore comprensione riportiamo, in figura, un esempio inerente i primi 4 passi di BLAST :
Se in una data posizione dell'allineamento l'aminoacido nella sequenza query e il corrispondente della sequenza del database coincidono, la lettera che lo rappresenta viene ripetuta nella riga tra le due sequenze.
Il carattere "+" indica che, nella corrispondente posizione dell'allineamento, l'aminoacido nella sequenza query e il corrispondente della sequenza del database sono simili.
Quando nella riga tra le due sequenze non è presente nessun carattere significa che, in quella posizione dell'allineamento, l'aminoacido nella sequenza query e il corrispondente della sequenza del database sono diversi, il carattere "-" in una delle due sequenze stà ad indicare la presenza di un gap.

5) Gli HSP con punteggio ( S o Score ) inferiore ad una certa soglia ( scelta empiricamente ) vengono scartati. L'output di BLAST conterrà le sequenze del database i cui HSP non sono stati scartati con i relativi punteggi ottenuti.
6) BLAST generalmente opera su database che contengono milioni di sequenze, questo fa sì che non tutti gli allineamenti prodotti abbiano una rilevanza biologica, ovvero non tutte le sequenze in output saranno omologhe alla sequenza query.
Dunque, è necessaria una valutazione statistica degli allineamenti prodotti da BLAST.
BLAST aggiunge ad ogni sequenza presente nell'output il suo E-Value, valore che, opportunamente interpretato dal ricercatore, indicherà quanto è probabile che il punteggio S indichi una correlazione biologica fra le due sequenze.
Analisi del risultato di BLAST :
Riportiamo di sueguito l'output di una ricerca effettuata con BLAST presso il sito dell'NCBI ( National Center For Biotechnology Information - Instituto che gestisce un database biologico pubblico ) e la sua analisi.
La pagina dei risultati di BLAST ( Fig.1 ) include un request ID ( RID - unico per ogni ricerca ) che identifica la ricerca effettuata, informazioni relative alla sequenza query, informazioni relative al database usato per la ricerca, una rappresentazione grafica ( Fig.2 ) del risultato della ricerca, la descrizione delle sequrenze che producono allineamenti significativi ( Fig.3 ) e gli allineamenti fra la sequenza di input e le sequenze presenti nel database ( Fig.4 ).

La rappresentazione grafica del risultato ( Fig.2 ) va interpretata in base a quanto segue :

Segue nella pagina di output ( Fig.3) l'elenco delle sequenze nucleotidiche del database scelto che producono allineamenti significativi con la sequenza query. Le sequenze sono ordinate in base all'E value (riquadro contrassegnato con 3 in figura 3 ), e in base al punteggio ( Score - riquadro contrassegnato con 2 in figura 3 ) ottenuto nell'allinemento, cliccando sul punteggio si visualizza l'allineamento della sequenza del database con la sequenza di input ( Fig.4 ). Ciascuna sequenza contiene un link ( riquadro contrassegnato con 1 in figura ), da cui si arriva alla entry di Entrez ( database contenente informazioni relative alle sequenze ) relativo a quella sequenza. A destra, per una data sequenza è inoltre eventualmente presente il link a LocusLink ( L ) , database contenente informazioni sulle sequenze ( riquadro contrassegnato con 4 in figura )

L'ultima parte dell'output di BLAST è occupata dagli allineamenti fra la sequenza input e le sequenze tratte dal database ( di cui riportiamo un esempio in Fig.4).
Per ciascun risultato è indicato prima dell'allinemento vero e proprio :
