L'allineamento di sequenze è il processo di appaiamento delle regioni simili condivise da due sequenze. Il nostro scopo è quello di associare ad ogni nuova sequenza una o più specifiche funzioni.
Lo scopo dell’allineamento di sequenze è quello di permettere al ricercatore di determinare se le sequenze da comparare dimostrano sufficiente somiglianza ( similarità )
per giustificare l’inferenza di omologia.
Si dicono omologhe quelle sequenze che hanno un gene ancestrale comune ovvero che condividono un progenitore. Il grado di similaritŕ fra due sequenze puň essere misurato, l'omologia č un dato qualitativo.
La similaritŕ di due sequenze è una quantitŕ misurabile che puň essere espressa come, per esempio,
percentuale di identitŕ. Il fine di tale operazione e di sfruttare il principio secondo il quale sequenze omologhe spesso, ma non sempre, hanno funzioni
comuni.
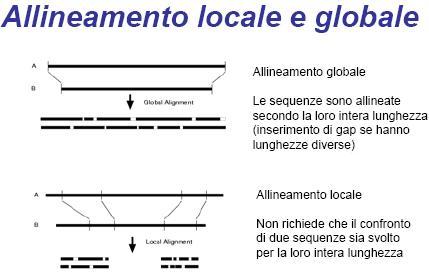
Si possono produrre due tipologie di allineamento, uno globale che prevede l'allineamento delle sequenze secondo la loro intera lunghezza, ed uno locale che non richiede che il confronto delle due sequenze sia svolto per l' intera lunghezza delle sequenze. Spesso si trova che alcune regioni di una proteina, o alcuni specifici aminoacidi, sono piu’ conservati di altri; questa informazione puň essere indicativa dei residui che sono piu’ importanti per il mantenimento della struttura o della funzione. Se perň partiamo da organismi strettamente correlati, come il topo ed il ratto, queste similaritŕ possono essere semplicemente indicative del fatto che una determinata regione non ha avuto ancora il tempo di divergere. I metodi di allineamento globale non tengono in considerazione questo fatto, e’ allora meglio utilizzare un sistema di allineamento che possa produrre un allineamento locale.
La figura mostra graficamente la differenza tra l'allineamento locale e l'allineamento globale
La prima semplice misura di similarità è data dalla somma del numero di appaiamenti di caratteri nelle due sequenze ( caratteri uguali nella stessa posizione nelle due sequenze ) .

In generale, per confrontare due sequenze di lunghezza, rispettivamente m ed n, il numero degli allineamenti generati N č pari alla somma della lunghezza delle sequenze meno 1 :
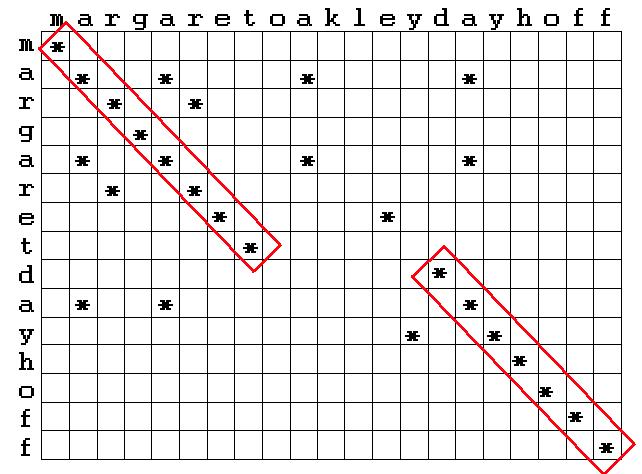
L'esempio riportato nella figura sottostante stigmatizza l'elevato numero di confronti necessari ( evidenziati all'interno dei rettangoli ) per calcolare la similarità di sequenza anche con sequenze molto brevi, inoltre nell'esempio non si tiene conto della possibilità di introdurre gaps ( inserzioni, cancellazioni ) in una delle due sequenze, cosa che aumenterebbe ancor più il numero di operazioni necessarie per il calcolo della similarità.

Quando si deve effettuare una ricerca per similaritŕ di sequenza in una banca dati, l’operazione di confronto tra due sequenze deve inoltre
essere ripetuta per ogni coppia di sequenze:
1) sequenza in input (query sequence) ;
2) ognuna delle sequenze della banca dati .
Tutto questo senza considerare le ulteriori complicazioni legate alla possibile presenza di gaps nell’allineamento tra due sequenze.
Come mostra l'esempio di seguito, vi č necessitŕ di trattare l’inserimento o la cancellazione di caratteri nell'allineamento (gaps), in quanto tale procedura ci porta generalmente ad allineamenti con punteggi più elevati.

L’apertura di un gap in una struttura proteica comporta uno stress che in realtŕ dipende soprattutto dalla regione strutturale in cui avviene l’inserimento o la delezione. Inserzioni e cancellazioni sono fortemente penalizzate all’interno delle regioni di struttura secondaria (α-eliche e filamenti β), in quanto costringono alla modifica delle interazioni con le strutture secondarie circostanti. Inoltre l’apertura di un gap non puň venire penalizzata con un punteggio negativo come ogni sua estensione, infatti una volta che la sequenza abbia accettato una inserzione o una cancellazione, la penalizzazione non deve essere necessariamente legata alla sua lunghezza.
Esempio:
Se la penalizzazione che imponiamo per l’apertura di un gap fosse il punteggio negativo -1, non vorremmo che l’inserzione di un residuo all’interno
di un loop fosse penalizzata di un punto e quella di 5 residui nella stessa posizione fosse penalizzata di 5 punti.
E' noto infatti che esistono diversi casi in cui ci possono essere inserzioni piů o meno lunghe in posizioni corrispondenti di un loop (vedi il caso
dei loops ipervariabili degli anticorpi) .
Un algoritmo di allineamento che tenga conto del possibile inserimento di un gap in ogni possibile posizione delle due sequenze e di ogni possibile
lunghezza di un gap in ogni possibile posizione sarebbe estremamente lento.
Gli allineamenti e il calcolo della similarità potrebbero essere notevolmente migliorati dall’introduzione di schemi di punteggio diversi da 0 e da 1 per l’appaiamento di residui amminoacidici; ad esempio si potrebbero prevedere punteggi alti per l’identità tra coppie di residui, punteggi un po’ più bassi ma maggiori di 0 per residui simili dal punto di vista chimico-fisico, punteggi invece negativi (o uguali a 0) per residui diversi o molto diversi dal punto di vista chimico-fisico. Sarebbe quindi utile definire una matrice di sostituzione ovvero una tabella che associ un valore ad ogni coppia di caratteri.
Es.
Nella figura sottostante vengono allineate le sequenze AAKKQW e AAKQW, nella riga fra le due sequenze vengono riportati i punteggi relativi agli accoppiamenti (A nessun carattere, AA, KA, KK, QQ, WW ) provenienti dalla matrice di sostituzione PAM 10, la somma di questi ( pari a 3 ) è il punteggio di similarità dell'accoppiamento.
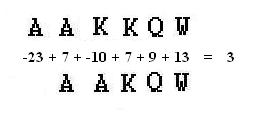
Quindi per effettuare un allineamento č prima di tutto necessario scegliere una matrice di sostituzione per valutare gli appaiamenti tra residui e definire
dei punteggi di penalizzazione per i gaps, ci si può inoltre avvalere di algoritmi di allineamento che utilizzano una tecnica di programmazione dinamica come quello di Needleman e Wunsch (1970)[1], oppure quello di
Smith e Waterman (1981)[2].
[1] Temple F. Smith and Michael S. Waterman ( 1981 ). Identification of Common Molecular Subsequences, J. Mol. Biol., 147:195-197.
[2] Needleman, S. B. & Wunsch, C. D. ( 1970 ). A general method applicable to the search for similarities in the amino acid sequence of two proteins. J.Mol. Biol. 48, 443- 453.