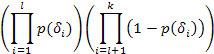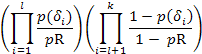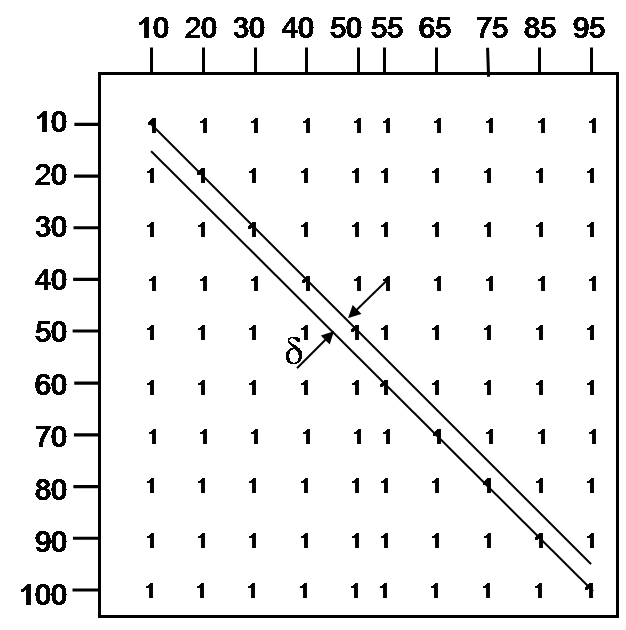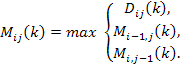


Introduzione
|
Il sequenziamento del patrimonio genetico di alcuni
organismi, principalmente quello umano, assieme
allo
sviluppo e ai progressi nei metodi e nelle
tecnologie di analisi, hanno aperto nuovi
scenari conferendo alle proteine un ruolo
sempre più importante suscitando un
interesse sempre maggiore presso la comunità
scientifica internazionale rendendo necessaria la nascita di un’ontologia che
permettesse di riferirsi al nuovo campo di ricerca:
la proteomica.
Le prima sezione presenta al lettore per grandi linee
cosa si accingerà a leggere e come è strutturato
complessivamente l’elaborato, la seconda sezione è una
breve introduzione al mondo delle proteine che può
servire a chi si avvicina per la prima volta a questo
argomento. In fase di stesura sono state seguite le seguenti regole che è utile sapere per una corretta navigazione e comprensione del testo:
E possibile accedere direttamente alle varie sottosezioni dell’elaborato mediante il click sulle voci che si trovano sulla sinistra. Il sito è stato ottimizzato per una risoluzione 800*600, è possibile che con risoluzioni maggiori alcune immagini possano risultare non leggibili, per tali foto è possibile ottenere un ingrandimento cliccandoci sopra. E’ indispensabile attivare javascript. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Le proteine |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Introduzione alle proteine
Altre presentano il ferro, lo zinco, il fosforo.
La proteina è una sostanza fondamentale per gli organismi viventi, animali o vegetali
ed formata da parti più piccole chiamate amminoacidi.
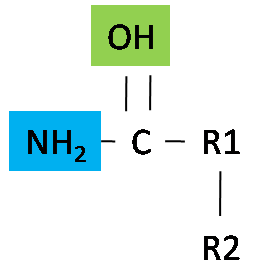
La composizione chimica di un amminoacido influenza la natura della proteina in quanto l’amminoacido è un suo composto essenziale. Gli amminoacidi si classificano in base ai gruppi "R" in:
Questo tipo di legame si forma tra il gruppo -NH2 e -COOH di due amminoacidi adiacenti. L’ insieme di più amminoacidi costituisce un peptide. Più peptidi formano un polipeptide ossia una proteina per cui polipeptide è un sinonimo di proteina. Talvolta può capitare di leggere davanti al nome di un amminoacido la lettera L oppure D ad indicare che quel amminoacido appartiene alla serie LEVOGIRA (ossia quelli che hanno il gruppo -NH2 sulla sinistra) oppure DESTROGIRA (quelli col gruppo -NH2 sulla destra) ad esempio: L-alanina oppure D-alanina. Le proteine possono contenere solo L-amminoacidi, quelli della serie D sono presenti in cellule di alcuni batteri; affinché si possano ottenere delle strutture stabili come quelle delle proteine è necessario che gli amminoacidi siano tutti della stessa serie. Le proteine sono tutte diverse tra loro. Si differenziano per:
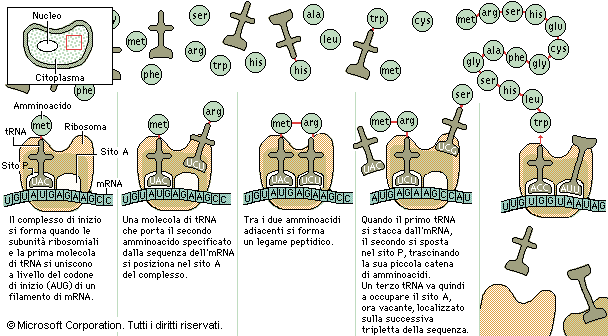
esse derivano da un lungo processo, chiamato sintesi proteica, che vede la partecipazione del Dna (acido deossiribonucleico), una molecola caratterizzata da unità fondamentale chiamata gene il quale è responsabile della trasmissione dei caratteri ereditari. Il compito del gene è quello di codificare un’intera sequenza di amminoacidi. Tuttavia un gene non costituisce direttamente una proteina, il collegamento tra proteine e geni è rappresentato dall’acido ribonucleico, o meglio conosciuto come RNA.
Per quanto riguarda la sintesi proteica è importante
sapere che essa è rappresentata da due fasi in particolare:
DNA-> RNA ->PROTEINA Una proteina viene classificata:
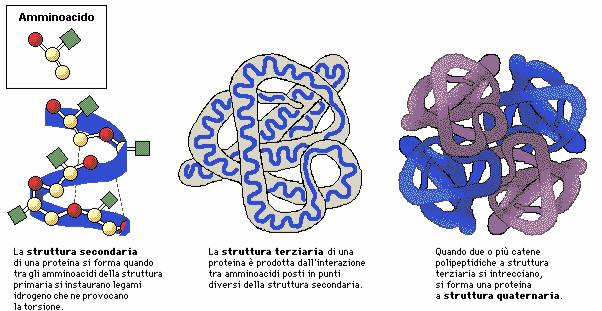
Struttura delle proteine
Esempi:
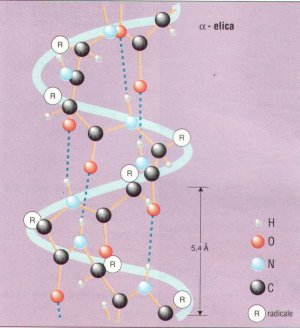
Esistono due varianti della struttura secondaria:
per riportarla nel quotidiano, l’elica è la tipica struttura della proteina che compone i nostri capelli.
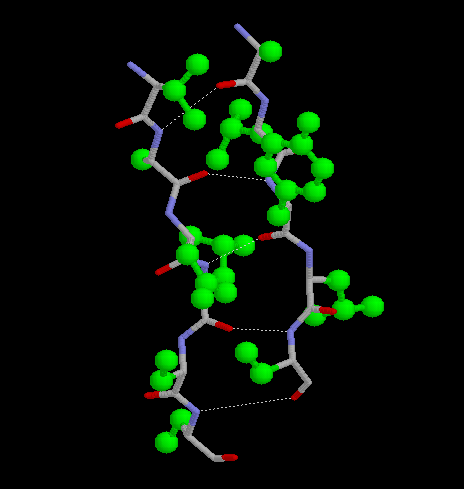
Le proteine sono strutturate in un certo modo tanto che si può parlare di conformazione, con tale termine si intente l’organizzazione strutturale che può subire modifiche senza che si abbia la rottura dei legami, tuttavia tra le tante forme che una proteina può assumere, una in particolare è quella maggiormente stabile dal punto di vista energetico. Questo tipo di conformazione viene detta nativa. Per giunta quando si parla di stabilità di una proteina ci si riferisce alla tendenza a mantenere la conformazione nativa. Un esempio di proteina stabile è l’alpha elica e il foglietto beta. Affinché una proteina conservi la sua stabilità e quindi la sua conformazione nativa sono necessarie una serie di interazioni chimiche, chiamate interazioni deboli, che essendo tantissime si sommano, predominano e stabilizzano la proteina.
Poiché la struttura di una proteina è legata
alla sua funzione ciò implica che un’alterazione
della struttura provocherà anche un’ alterazione
nella funzione che viene definita denaturazione.
Questo processo è reversibile perché tutte le informazioni necessarie di una struttura proteica sono contenute nella sequenza di amminoacidi. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tecniche di laboratorio |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Introduzione
La prima tecnica si pone l’obiettivo di separare le proteine che compongono l’elemento da analizzare al fine di poterle poi successivamente identificare con una delle varie tecniche quali la cromatografia (nelle sue diverse varianti) o spettrometria di massa (nelle sue varianti). Il sequenziamento automatizzato consente poi di determinare l’esatta sequenza delle proteine che compongono un composto.
Elettroforesi
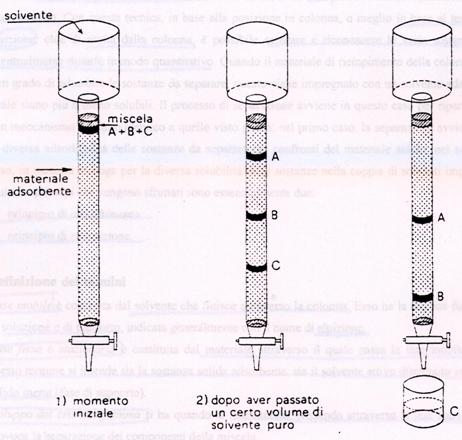
Cromatografia su colonna:
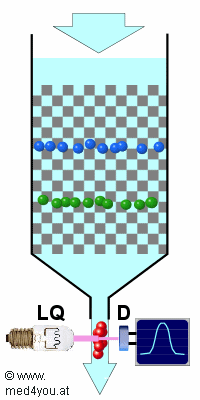
La cromatografia è un metodo che ci consente di
separare, identificare i componenti all’interno di
una miscela.
L’efficienza della colonna cromatografica viene indicata
dalla banda cromatografica, in
particolare:
L’allargamento della banda cromatografica può essere dato da una serie di fattori, tra cui:
HPLC
TLC
Cromatografia a scambio ionico
Spettrometria di massa (e cenni di spettroscopia)
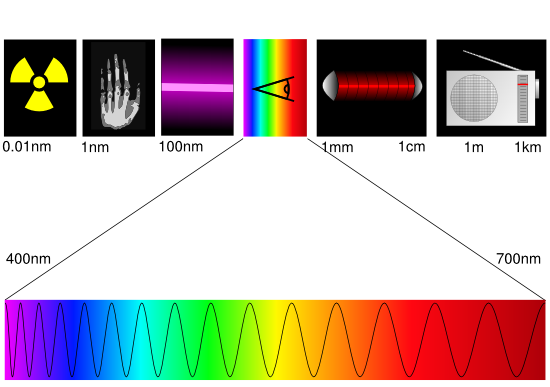
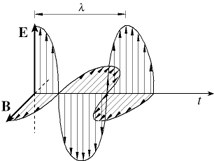
La spettroscopia sfrutta la caratteristica delle molecole organiche ed inorganiche di assorbire la luce, questo è possibile grazie ad alcuni gruppi funzionali. Si deve premettere che determinati composti per poter assorbire nell’ultravioletto e nel visibile (UV\VIS) devono possedere una struttura elettronica eccitabile, mentre le regioni ultraviolette e visibili devono possedere l’ energia necessaria per poter spostare gli elettroni di questi composti. La struttura eccitabile è affidata proprio a quei gruppi funzionali, di cui sopra, chiamati cromofori. L’eccitazione di questi elettroni, vengono studiate attraverso gli spettri di assorbimento. Lo spettro di assorbimento è un diagramma cartesiano, in cui sono messe a paragone un fascio di radiazioni che diminuisce di intensità, con i vari parametri di un’onda elettromagnetica. Un’onda elettromagnetica è rappresentata dalla:
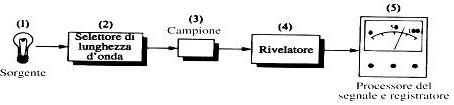
Lo strumento di cui ci si serve per poter misurare l’assorbimento della luce è chiamato spettrofotometro. Nel nostro caso i risultati che ci vengono forniti servono ad identificare e valutare la concentrazione delle nostre proteine in una soluzione. Quando facciamo passare un fascio di luce attraverso una soluzione di concentrazione "c" e di spessore "b" l’intensità del fascio di luce diminuisce perché essa viene assorbito. Questa relazione è rappresentata della Legge di Lambert e Beer: essa presuppone che la luce incidente sia parallela e monocromatica e che la concentrazione della soluzione sia bassa, poiché ad alte concentrazioni si ha un avvicinamento tra le particelle, che genera delle interazioni che sono in grado di modificare la capacità di assorbimento ad una data lunghezza d’onda. A= a*b*c dove "A" sta per attenuazione del raggio. Quello che abbiamo fino ad ora descritto è un modello di uno spettrofotometro:
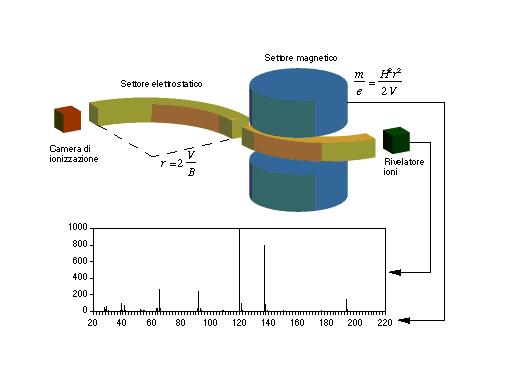
Per la spettroscopia di massa gli analiti vengono dapprima ionizzati in fase gassosa in modo che introdotti in un campo elettrico possano raggiungere una certa velocità, questa operazione può essere effettuata mediante diverse tecniche: l’espulsione di elettroni (genera uno ione-radicale, ossia una molecola caricata elettronicamente che presenta un solo elettrone spaiato rendendo la particella estremamente reattiva), la protonazione, la deprotonazione, la cationizzazione. Le molecole così ionizzate sono instabili e si frammentano in ioni più leggeri secondo schemi tipici in funzione della loro struttura chimica, le molecole caricate vengono poi esaminate attraverso un analizzatore cioè entrano in un dispositivo capace di separare gli ioni in funzione del loro rapporto massa/carica (m/z), infine viene utilizzato un rilevatore: si tratta generalmente di dinodo, cioè un moltiplicatore elettronico capace di amplificare la debolissima corrente prodotta dagl’ioni che hanno superato l’analizzatore. I segnali ottenuti in questo modo vengono poi trasmessi ad un calcolatore in grado, con l’opportuno software, di rappresentare l’abbondanza di ogni ione in funzione della sua massa, cioè lo spettro di massa finale. Gli spettri vengono normalmente rappresentati come istogrammi che riportano l’abbondanza di ogni ione in funzione della sua massa, ipotizzando ragionevolmente che tutti gli ioni prodotti dall’analisi abbiano carica singola. Le abbondanze vengono riportate come rapporto rispetto al picco base, che è il picco più abbondante osservato nello spettro. Tale normalizzazione permette di avere spettri che sono funzione solamente dell’analita e delle condizioni di analisi. Il picco base non sempre coincide con il picco genitore, che è invece il picco che corrisponde alla molecola ionizzata e che consente di stabilire quindi il peso molecolare dell’analita. In genere, più uno ione molecolare è stabilizzato (per effetto induttivo o per risonanza, maggiore è la sua probabilità di giungere intatto al rivelatore, maggiore quindi sarà la sua abbondanza. Dall’abbondanza del picco genitore è possibile già ipotizzare a quale classe di composti appartenga l’analita. Nel caso della ionizzazione da impatto a medie energie si ha:
Qualora si desideri aumentare l’abbondanza del picco genitore, occorre o ridurre l’energia impiegata nella ionizzazione da impatto o ricorrere a tecniche di ionizzazione più soft quali la ionizzazione chimica o di campo. L’immagine seguente tenta di schematizzare tutto il processo della spettroscopia di massa:
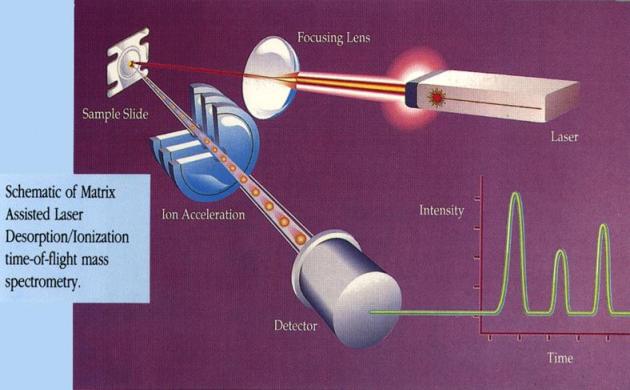
C’è da aggiungere che questa tecnica inizialmente non veniva applicata alle proteine poiché le misure di m/z venivano effettuate nella fase gassosa e siccome sia il riscaldamento sia gli altri trattamenti per portare la molecola in fase gassosa erano in grado di decomporla si è preferito introdurre due nuove tecniche per ovviare al problema:
Nella prima tecnica le proteine sono poste in una sostanza, come il glicerolo, che assorbe luce (chiamata matrice). Con un impulso breve di luce laser (luce UV), le proteine si staccano da questa matrice ed aumentano la loro velocità e sono rilasciate in sistema sotto vuoto, che rappresenta un analizzatore. Questo metodo è molto utilizzato per studiare la massa di moltissime macromolecole.
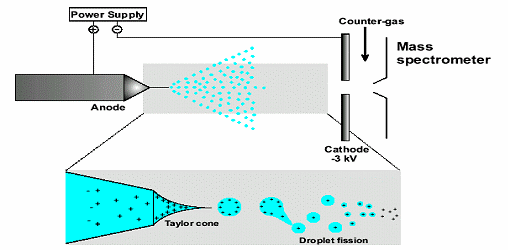
Per quanto riguarda la seconda tecnica: la miscela di analiti passa attraverso un ago carico mantenuto ad alto potenziale elettrico e viene disperso sotto forma di micro gocce cariche. Il solvente dalle gocce evapora e gli ioni delle macromolecole che si sono prodotti passano in fase gassosa, senza subire danni. Tuttavia i protoni aggiunti durante il passaggio attraverso l’ago procurano un’aggiunta di cariche alle proteine, il rapporto massa /carica lo si può analizzare in camera da vuoto.
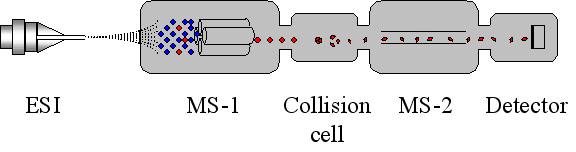
La spettrometria di massa richiede piccolissime quantità di materiale e quindi può essere applicata a piccole quantità di un campione estratto mediante elettroforesi. Uno dei parametri per poter identificare una proteina è la massa, per cui una volta che la si è calcolata attraverso la spettroscopia si possono considerare i vari cambiamenti. La spettrometria di massa può essere utilizzata per sequenziare frammenti di amminoacidi piuttosto brevi, quindi è un’ottima tecnica di identificazione di una proteina. Un’importante tecnica, in questo senso, è TANDEM MS o meglio conosciuta come MS/MS: la proteina viene pretrattata con un reagente chimico per ottenere vari frammenti, la miscela viene iniettata in uno strumento che altro non è che due spettrometri posti in serie.
Lo spettrometro è costituito da una camera MS-1 che
ha come compito quello di selezionare tra i vari
ioni quello desiderato.
Sequenziamento automatizzato (metodo di Edman e Sanger)
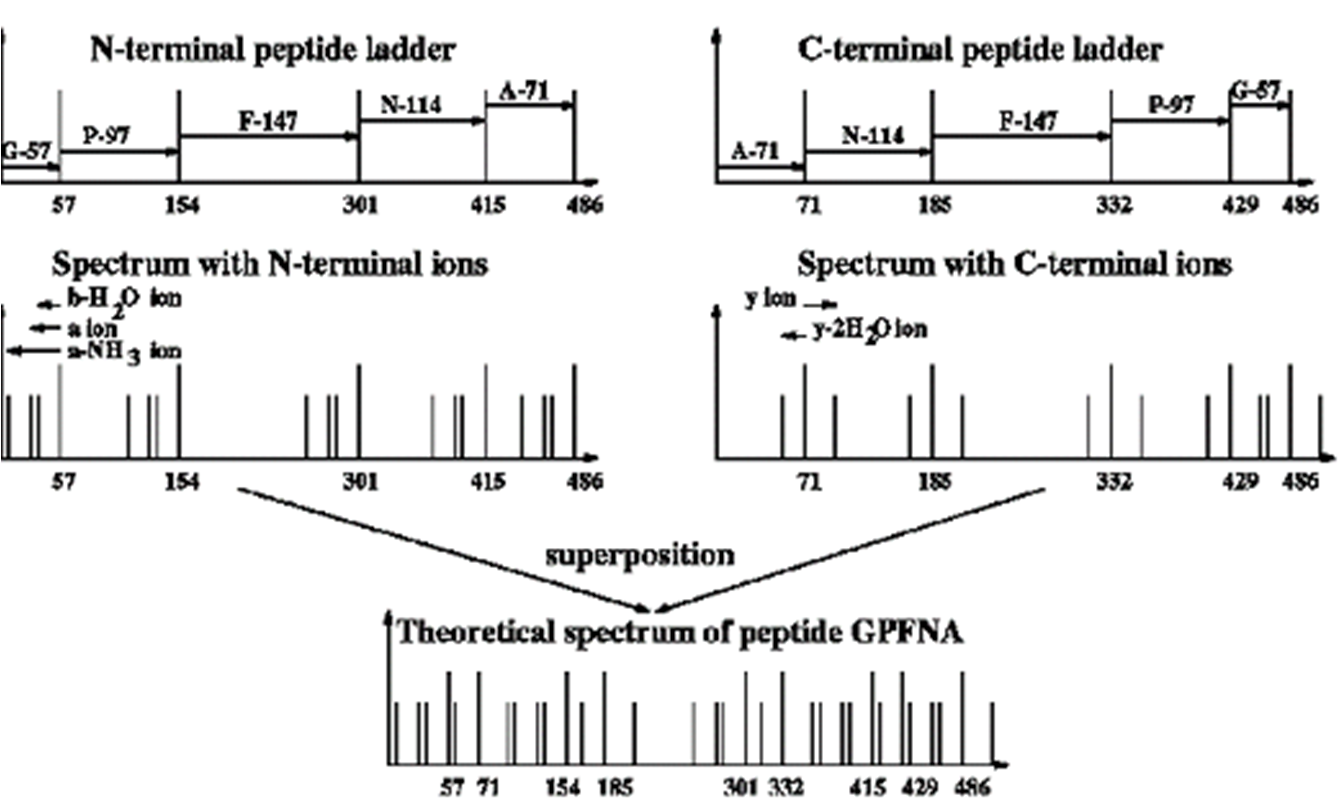
Per quanto riguarda la composizione amminoacidica, essa è diversa per ogni proteina per cui può essere utilizzata una specie di impronta digitale (fingerprint) che serve a stabilire ad esempio se il tipo di proteine isolate in laboratori differenti sono o meno le stesse. Tuttavia l’idrolisi (reazione chimica causata da una molecola d’ acqua nella quale si verifica la scissione della proteina in tanti aminoacidi o peptidi) di una sola proteina non è sufficiente per poter determinare la sequenza amminoacidica di un intero complesso, per cui si procede con una seconda tecnica che è quella dell’identificazione di -NH2. Questo tipo di identificazione si basa su di una tecnica di marcatura del residuo -NH2 (chiamato N-terminale) con dei reagenti. Attraverso questi reagenti il polipeptide viene idrolizzato (scisso in parti più piccole) e si ottiene l’identificazione degli amminoacidi marcati. Purtroppo questa metodica ha una limitazione:a causa dell’idrolisi il polipeptide viene distrutto, per cui è applicabile una sola volta di conseguenza non può identificare i residui successivi all’ N-terminale. Per ovviare a questo tipo di inconveniente è stata utilizzata un’altra tecnica, in cui si marca e stacca soltanto il residuo -N terminale, lasciando intatta l’intera catena. A differenza della precedente tecnica, questa può essere usata più volte, ed è sfruttata da uno strumento detto sequenziatore che è una macchina in grado di mescolare i reagenti, separare i prodotti ed infine registrare i risultati. Nell’eventualità che ci trovassimo di fronte a delle proteine di grandi dimensioni, sarà necessario prima staccare i singoli frammenti attraverso dei metodi chimici e poi sequenziare e purificare ogni singolo frammento ottenuto con il sequenziatore. Dopo tale procedura si prosegue nella determinazione dell’ordine in cui i frammenti sono stati disposti nella proteina di partenza: si prende in considerazione un altro campione della stessa proteina, che subisce lo stesso sequenziamento ma i punti di rottura della proteina saranno differenti rispetto al primo campione. In questo modo è possibile esaminare i frammenti ottenuti con le due rotture con la possibilità di sovrapporli e cercando di riscontrare dei punti di contatto. Così facendo si ottiene l’esatta combinazione dei peptidi ottenuti dalla prima rottura. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Problemi computazionali |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Il problema del Sequenziamento
di un peptide
Durante
questa fase può accadere che si perdano delle
piccole parti e conseguentemente il frammento
risulterà di massa più bassa..
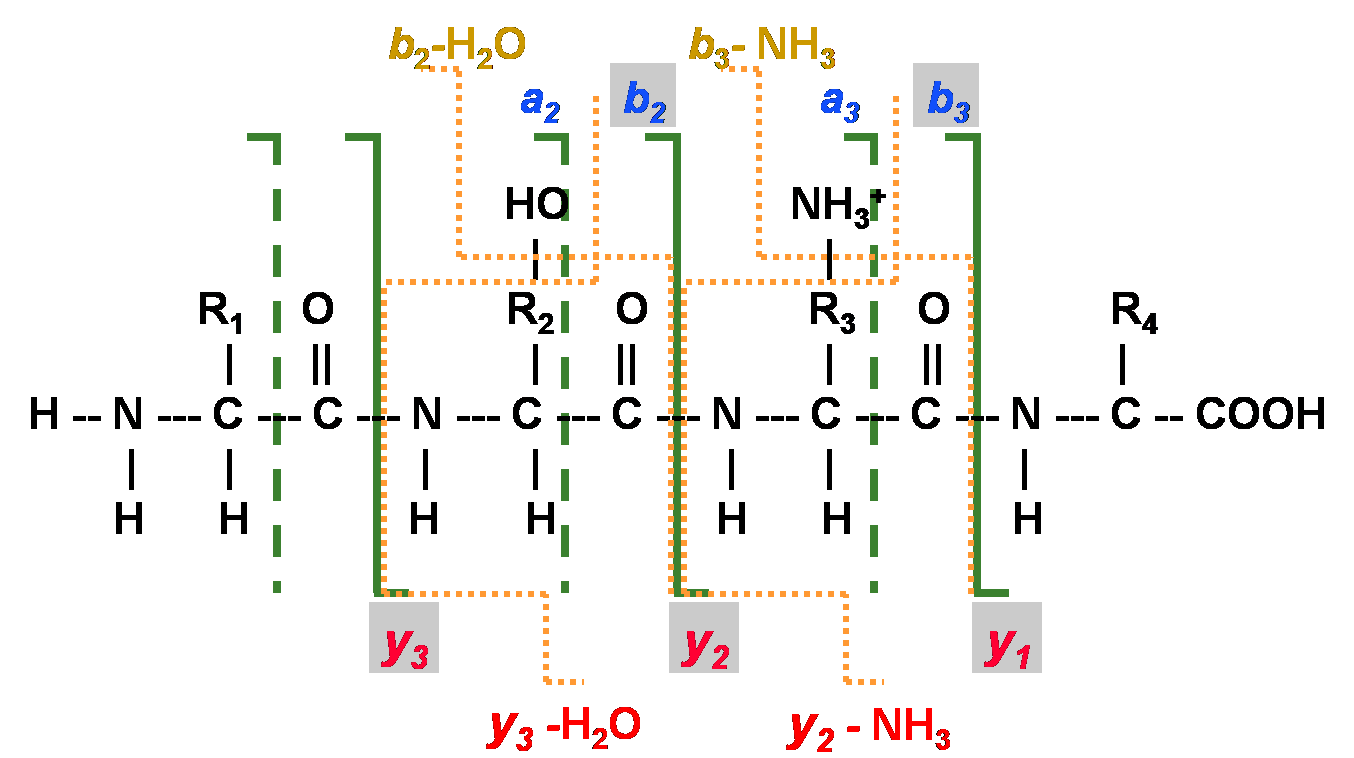
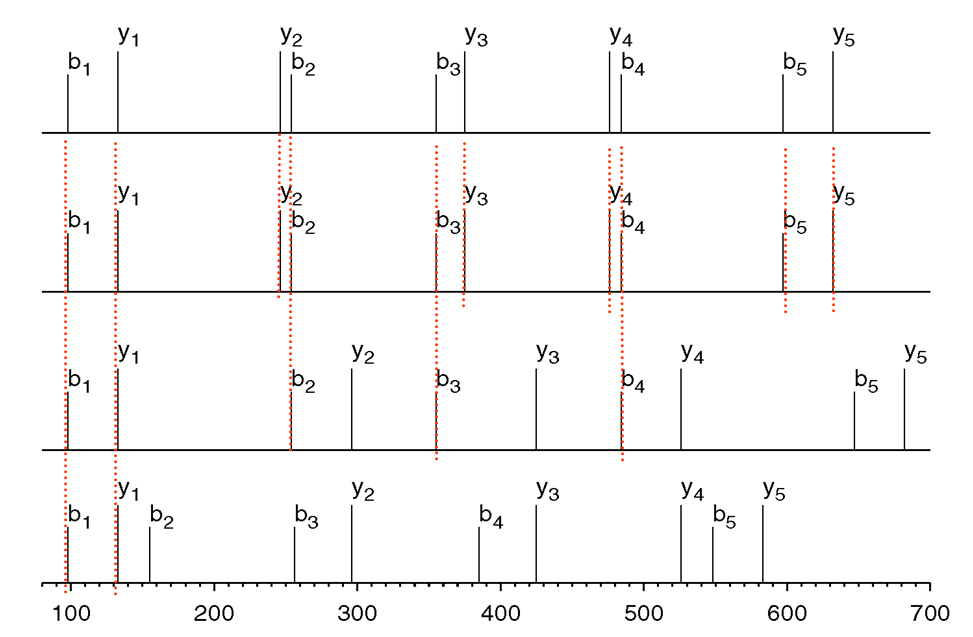
Per quanto riguarda lo spettrofotometria di massa tandem, lo spettro teorico T(P) di un peptide P può essere calcolato sottraendo tutti i possibili ioni δ1, … , δk dalle masse di tutti i parziali peptidi di P cosicché ogni peptide generi k masse nello spettro teorico:
Uno spettro sperimentale S ={s1,
..., sq} è un insieme di numeri
ottenuti da un esperimento (di spettrometria di
massa) comprendendo anche le masse di alcuni
ioni di frammenti e il rumore (distorsioni,
disturbi) chimico. Distinguiamo lo spettro
teorico T(P) da quello sperimentale S
in quanto viene generato matematicamente a
partire dalla sequenza del peptide P,
contrariamente S viene generato
sperimentalmente senza sapere quale sia la
sequenza peptidica che l’abbia generato. .
I punti di coincidenza tra lo spettro
sperimentale S e il peptide P
esprime il numero di masse in S che
sono uguali alle masse in T(P),
questo numero viene chiamato shared peaks
count (SPC).
In
realtà lo spettrometro di massa misura la massa
e l’intensità
rispecchiando il numero di ioni di frammenti di
una data massa rivelati dallo spettrometro e di
conseguenza gli spettrometristi di massa
rappresentano lo spettro in due dimensioni
parlando di masse nello spettro come picchi.
Il
primo approccio implica la generazione di
tutte le 200l sequenze di
aminoacidi di lunghezza l e i propri
spettri teorici corrispondenti, con
l’obiettivo di trovare una sequenza con il
miglior matching tra gli spettri sperimentali
e gli spettri teorici della sequenza. Dato
che il numero di sequenze cresce
esponenzialmente con la lunghezza del
peptide, sono state progettate diverse
tecniche (di tipo branch-and-bound) per
limitare l’impennata combinatoria in questi
metodi. La tecnica del
prefix pruning
(taglio del prefisso) restringe lo spazio
computazionale alle sequenze i cui prefissi
coincidono bene con lo spettro sperimentale.
La tecnica del
prefix pruning ha il problema che frequentemente scarta la
sequenza corretta se i suoi prefissi sono
scarsamente rappresentati nello spettro.
Comunque ci sono probabilità inerenti a rumore chimico, cioè esso può produrre qualsiasi massa (che non ha niente a che fare un l’interesse per un peptide) con una certa probabilità pR. Perciò bisogna aggiustare il valore probabilistico come:
Ad esempio per k = 4 e assumendo che per un peptide parziale P’ si vedano solo gli ioni δ1, δ2 e δ4, il valore calcolato sarà:
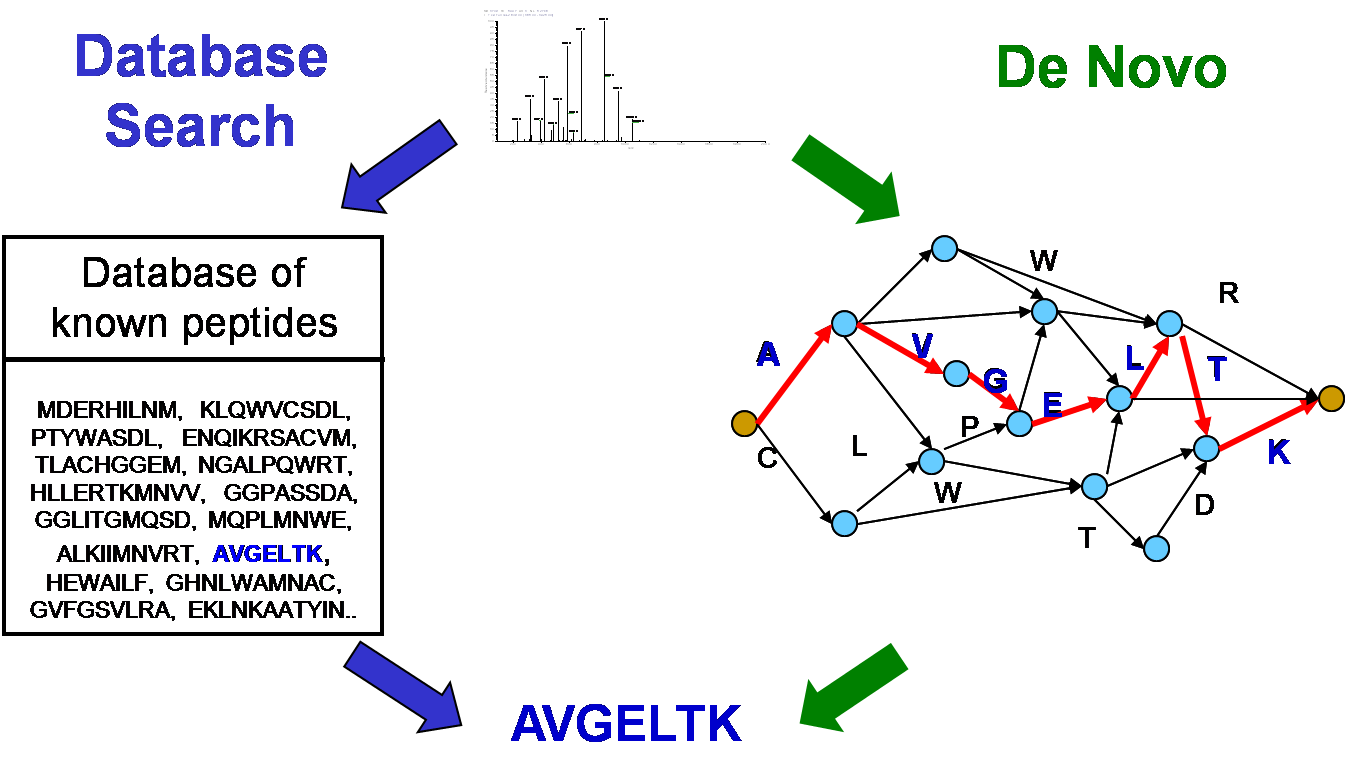
Identificazione delle proteine
mediante la ricerca su database
L’approccio di base dell’algoritmo SEQUEST è una ricerca lineare attraverso il database. Spesso i peptidi in una cellula sono leggermente differenti dal “canonico” peptide presente nel database e ciò rappresenta un ostacolo per gli algoritmi di ricerca (come il SEQUEST) su database di tipo MS/MS. C’è da dire che per una proteina la sintesi su un ribosoma non è il passo finale nel suo ciclo di vita, infatti molte proteine sono soggette a ulteriori modifiche che consentono di regolare le proprie attività e tali modifiche possono essere sia permanenti che reversibili. Per esempio, l’attività enzimatica di alcune proteine è regolata attraverso la rimozione o l’aggiunta di un gruppo fosfatico (serina, tirosina, treonina) ad un specifico residuo, ciò accade con la fosforilazione che è un processo reversibile: la proteina chinasi aggiunge il gruppo fosfatico invece la fosforilasi rimuove tale gruppo.
Le proteine sono formate da un sistema complesso
necessario al signaling cellulare e la regolazione
metabolica e sono perciò spesso soggette a un vario
numero di modifiche biochimiche (per esempio la
fosforilazione o la glicolisazione). Infatti, quasi
tutte le sequenze delle proteine sono modificate
dopo essere state costruite dal loro modello di mRNA,
si conosco 200 tipi di modifiche dei residui degli
aminoacidi. Siccome siamo impossibilitati nel
prevedere queste modifiche post-traduzione da una
sequenza di un DNA, la ricerca delle modifiche che
avvengono rimane un importante problema aperto.
La maggiore difficoltà che si ha con il problema
dell’identificazione di proteine modificate è che
peptidi molto simili P1
(il peptide presente nel database) e
P2 (il peptide corrispondente alle
versione modificata di P1)
possono avere spettri S1 e
S2 molti differenti.
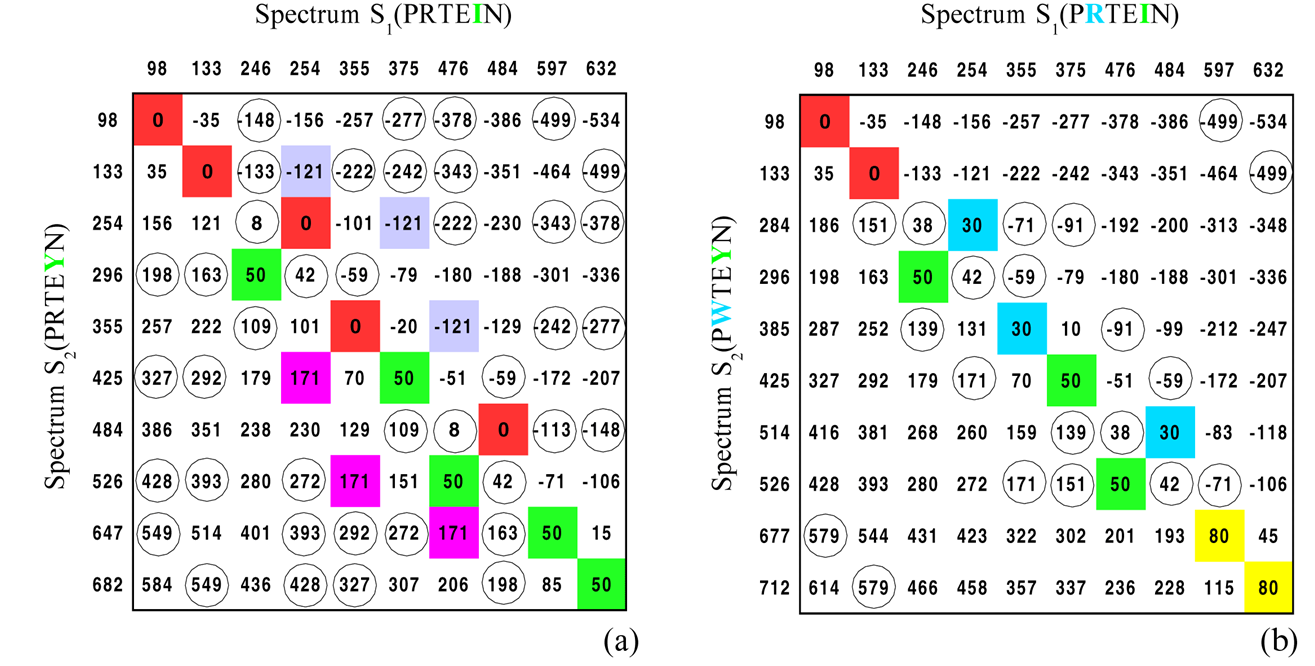
L’immagine di sopra dimostra l’inefficacia dell’utilizzo
del SPC, senza mutazioni l’SPC è pari a 10 (per la
proteina PRTEIN il cui spettro è pari a {98, 133,
246, 254, 355, 375, 476, 484, 597, 632}) , con una
singola mutazione il valore di SPC è 5 (PRTEIN
mutata in PRTEYN con spettro uguale a {98, 133, 254,
296, 355, 425, 484, 526, 647, 682}) infine per due
mutazioni il valore di SPC scende a 2 (PRTEIN mutata
in PGTEYN il cui spettro {98, 133, 155, 256, 296,
385, 425, 526, 548, 583}).
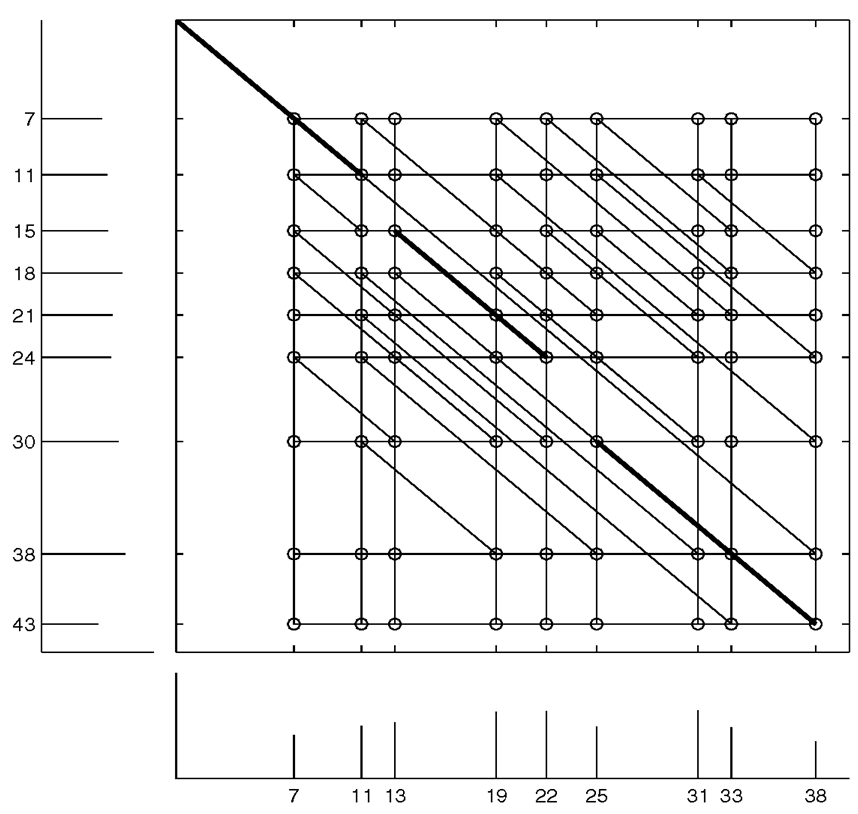
La convoluzione spettrale
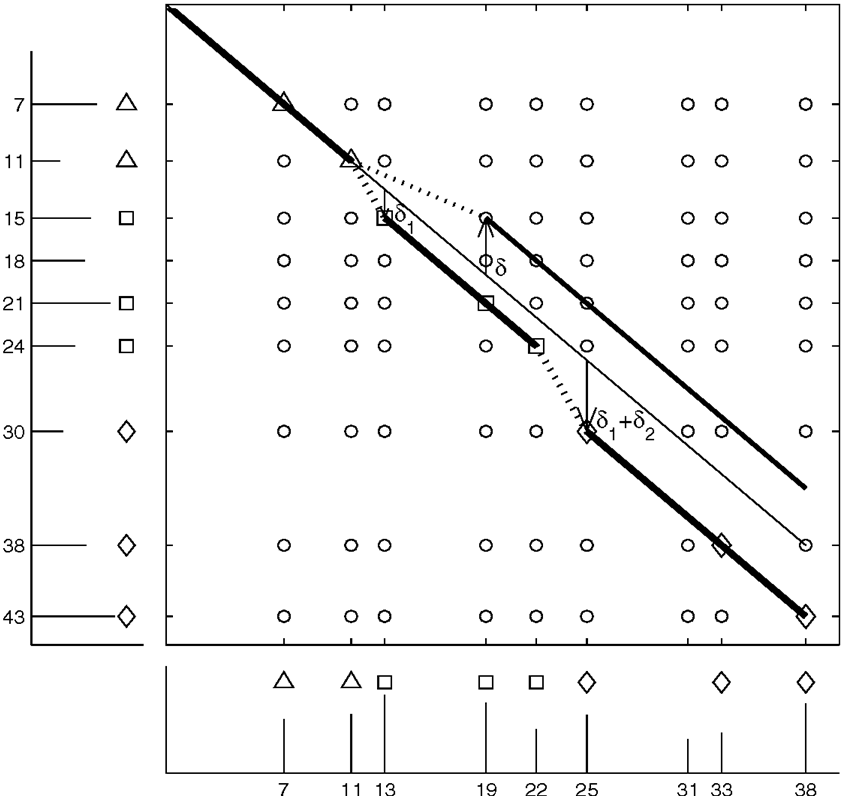
La figura 25 mostra gli elementi della convoluzione
spettrale S2ӨS1 rappresentati come elementi di una matrice di differenza.
S1
e S2 sono rispettivamente gli
spettri teorici dei peptide PRTEIN e
PRTEYN. Gli elementi nella convoluzione
spettrale aventi molteplicità più grande di 2 sono
colorati, invece gli elementi aventi molteplicità
pari a 2 sono cerchiati.
L’SPC introdotto nella sezione precedente indica il
numero di masse comuni a S1
e a S2 ed è semplicemente
(S2ӨS1)(0),
ritornando alla figura 25 sono colorati di
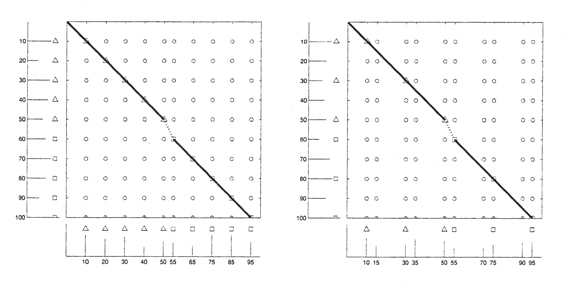
rosso.. S={10, 20, 30, 40, 50, 60, 70, 80, 90, 100} associato al peptide P (per semplicità assumiamo che
produca solo b-ioni). SI={10, 20, 30, 40, 50, 55, 65, 75, 85, 95} e SII={10, 15, 30, 35, 50, 55, 70, 75, 90, 95}
che sono gli spettri teorici presenti nel database
rispettivamente dei peptidi PI
e PII. A questo punto viene
da chiedersi quale dei due spettri sia il più
appropriato ad S. Attraverso l’uso del SPC non ci è
consentito rispondere a questa domanda perché sia
SI sia SII
hanno cinque picchi in comune con S.
Inoltre, anche la convoluzione spettrale non è in
grado di dare risposta a questa domanda siccome sia
SӨSI e SӨSII
indicano picchi forti della stessa altezza a 0 e a
5. Questo implica che entrambi PI
e PII possono essere
generati da P mediante una singola
mutazione con differenza di massa pari a 5.
Attraverso una analisi più approfondita si dimostra
poi che questa mutazione può avvenire introducendo
uno spostamento di 5 dopo la massa 50 solo per
PI e non per PII.
La differenza maggiore tra gli spettri SI
e SII sta nel fatto che per il primo le
posizioni in cui si ha la migliore coincidenza con
lo spettro S sono raggruppate. S={10, 20, 30, 40, 50, 60, 70, 80, 90, 100} in SI={10, 20, 30, 40, 50, 55, 65, 75, 85, 95} perciò D(1)=10 (dopo 1 spostamento 10 elementi sono in comune) per questi due insiemi. L’insieme SII={10, 15, 30, 35, 50, 55, 70, 75, 90, 95} ha cinque elementi in comune con S (gli stessi di SI) ma non c’è una la possibilità che un singolo spostamento trasformi S in SII (infatti con uno spostamento abbiamo 6 elementi in comune D(1)=6). Sotto analizzeremo e risolveremo il problema dell’Allineamento dello spettro:
Per primo possiamo rappresentare i due insiemi
A = {a1, …, an}
e B = {b1, …, bm}
come array contenenti 0 e 1 di nome a
e b di lunghezza pari a
an e bm.
L’array a conterrà n
1 (alla posizione a1, …, an)
e an-n 0, finché l’array
b non conterrà m 1
(alla posizione b1, …, bm)
e bm-m 0. In tale
modello, uno spostamento δi
< 0 è semplicemente l’eliminazione dei δi
0 in a. Tenendo presente questo
modello, il problema dell’allineamento spettrale
è semplicemente trovare la distanza di edit (il
numero di operazioni per trasformare a
in b o b in a)
tra a e b quando le
operazioni elementari sono l’eliminazione e
l’inserimento di blocchi di 0. Queste operazioni
possono essere modellate da tante punte
orizzontali e verticali in un grafico di tipo
Manhattan. Il problema dell’Edit Distance e
quello dell’allineamento spettrale si
differenziano per un alfabeto alquanto inusuale
e il tracciato del percorso nel grafico
risultante.
In entrambi i casi, il numero di 1 sulla diagonale principale è lo stesso, e D(0) = 5. Il δ-SPC è il numero di 1 sulla diagonale che è δ distante dalla diagonale principale. La limitazione della convoluzione spettrale è che essa considera le diagonali separatamente senza combinarle in un possibile scenario di mutazione. La k-similarità della convoluzione spettrale è definita come il numero massimo di 1 su un percorso lungo la matrice spettrale che usa al massimo k + 1 diagonali, e il k-allineamento spettrale ottimale è definito come il percorso che usa queste k + 1 diagonali. Per esempio, l’ 1-similarità è definita come il numero massimo di 1 sul percorso attraverso questa matrice che usa al più due diagonali. La figura 27 illustra il concetto di 1-similarità e mostra che S è assomiglia più a SI che a SII; nel primo caso il percorso bidimensionale copre dieci 1 (la matrice di sinistra) contro i sei del secondo caso (la matrice di destra).
La figura 28 mostra che l’allineamento spettrale rileva più e più similarità impercettibili tra spettri semplicemente incrementando di k, si vedrà che D(0) = 3, D(1) = 5 e D(2) =8.
L’allineamento spettrale spettrale
La k-similarità tra A e B è data da:
L’algoritmo sin qui esposto per l’allineamento
spettrale è piuttosto lento ed ha tempo di
esecuzione pari a O(n4k) per
due spettri di n-elementi.
Allora la ricorrenza per Dij(k) può essere scritta come:
La ricorrenza per Mij(k) è data da:
La trasformazione del grafo di programmazione dinamica può essere ottenuta attraverso l’introduzione di spigoli verticali ed orizzontali che forniscono la possibilità di commutare tra le diagonali.
Il punteggio (score) del percorso è il numero di 1
su questo percorso, mentre k corrisponde al
numero di commutazioni (il numero delle diagonali
usate meno 1). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Conclusioni |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Si è visto che le proteine sono composti organici tra i più complessi
costituenti fondamentali di tutte le cellule animali e vegetali.
Dal punto di vista chimico, una proteina è un polimero
(o anche una macromolecola) costituita da una combinazione
variabile di 20 diversi monomeri detti amminoacidi spesso
in associazione con altre molecole e/o ioni metallici (in questo
caso si parla di proteina coniugata).
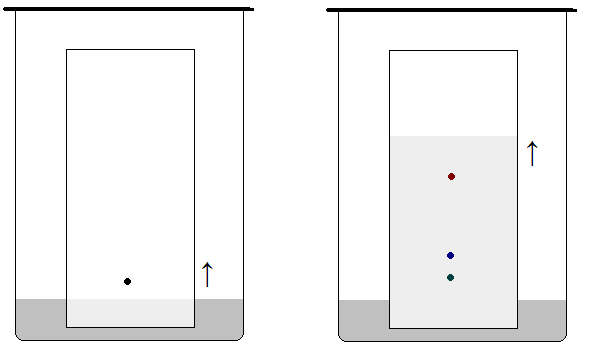
La cromatografia si basa sul fatto che i vari componenti
di una miscela tendono a ripartirsi in modo diverso tra due
fasi, in funzione della loro affinità con ciascuna di esse.
Mentre una fase rimane fissa (la fase stazionaria),
ed è generalmente un solido o un gel, un’altra fase, liquida
o gassosa, (la fase mobile) fluisce su di essa trascinando
con sé in quantità maggiore i componenti della miscela che
più risultano affini a lei.
Il primo è la delucidazione della sequenza della
proteina nel caso in cui un campione biologico
contenga una proteina che non è presente sul
database o differisce dalla versione canonica
presente sul database, il secondo è
l’identificazione di una proteina che è presente in un
database. Entrambi i problemi computazionali hanno
lo stesso obbiettivo, lo stesso input e lo stesso
output, la differenza principale tra i due è che il
secondo problema fa riferimento ad un database. |