L'analisi comparativa rappresenta certamente l'approccio bioinformatico più rilevante per la caratterizzazione funzionale delle sequenze nucleotidiche e proteiche. Difatti, il modo più efficace per decifrare un testo scritto in una lingua sconosciuta è quello di confrontarlo con testi equivalenti scritti in linguaggi noti. Per esempio, in questo modo è stata possibile, in seguito al ritrovamento della stela di Rosetta che riportava il testo di un decreto del 196 a.C. in tre diverse lingue compreso il greco, la decifrazione dei caratteri geroglifici.
In modo analogo si può predire la funzione di un gene o di una proteina sulla base dell'osservazione di somiglianza o similarità significativa con altri geni o proteine a funzione nota. Inoltre, si possono valutare le proprietà funzionali di ciascuno dei siti che compongono un gene o una proteina sulla base del confronto di un certo numero di sequenze simili, relative allo stesso o a diversi organismi, che come si vedrà possono anche essere definite omologhe.
Per poter effettuare analisi comparative è necessario selezionare un certo numero di sequenze omologhe e costruire successivamente un "allineamento multiplo".
L’allineamento multiplo di tre o più sequenze può essere definito come un’ipotesi di omologia posizionale tra basi o aminoacidi. Esso viene rappresentato sotto forma di una tabella costituita da righe, corrispondenti alle sequenze omologhe considerate, e da colonne, corrispondenti a ciascun sito dell’allineamento. Per ipotesi di omologia posizionale si intende che tutti i residui presenti nella stessa colonna di un multiallineamento siano evolutivamente correlati. Esso viene realizzato utilizzando gli algoritmi di allineamento globale, che quindi considerano l’intera lunghezza delle sequenze in esame.
Al fine di trovare un’ accettabile soluzione al problema sono stati proposti metodi approssimati basati sulla procedura di allineamento progressivo.
Algoritmi per l’allineamento multiplo:
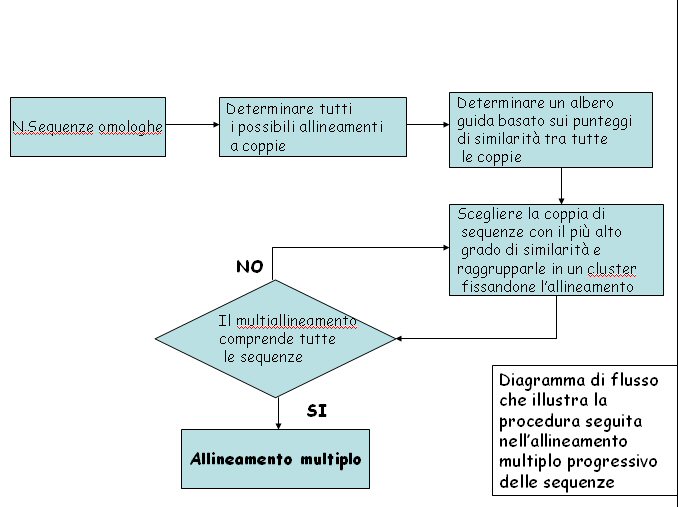
Il punto di partenza di un allineamento progressivo è l’allineamento tra tutte le possibili coppie di sequenze. Date N sequenze, si dovranno effettuare N(N-1)/2 allineamenti a coppie. I punteggi di similarità calcolati tra tutte le diverse coppie di sequenze possono essere utilizzati per costruire un albero, il quale verrà utilizzato come guida per l’allineamento multiplo progressivo. Questo viene effettuato formando via via dei cluster di sequenze allineate, costituiti da due o più sequenze il cui allineamento sia stato precedentemente fissato. Un cluster può essere allineato a una sequenza o ad un altro cluster mediante l’allineamento a coppie.
Vantaggio:
Molto veloce e consente l’allineamento multiplo di un gran numero di sequenze in un tempo molto limitato.
Svantaggio:
Se in una qualunque fase viene introdotto un errore nell’allineamento, questo errore si propagherà nelle fasi successive e non potrà più essere corretto.