Nel caso dell'allineamento di due sequenze di aminoacidi è opportuno applicare dei criteri di similarità che non si limitino a verificare l’identità assoluta, ma considerino anche che gli aminoacidi possano essere più o meno simili tra loro.
Le matrici di sostituzione rispondono a questa esigenza. Esse tipicamente comprendono 210 valori: 20 (sulla diagonale) relativi al punteggio da attribuire all’appaiamento di ogni aminoacido con se stesso, gli altri 190 relativi a tutte le possibili sostituzioni aminoacidiche (19 + 18 + ... + 2 + 1). Spesso i 190 valori sono riportati per comodità anche nella loro parte speculare, per esempio la relazione alanina-arginina è riportata sia come tale, sia come arginina-alanina. In tal modo queste matrici vengono rappresentate nel loro tipico formato simmetrico di 20 x 20 valori.
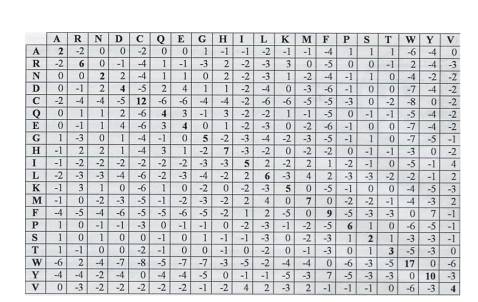
La figura rappresenta la matrice di sostituzione PAM240.
Questa matrice è spesso utilizzata per la ricerca di similarità in sequenze filogeneticamente distanti.
Con le matrici di sostituzione è quindi possibile attribuire un punteggio appropriato a ogni coppia di aminoacidi appaiati in un allineamento, piuttosto che contare semplicemente il numero di identità. Ma come vengono create queste matrici? Cosa rappresentano? E quale matrice di sostituzione è più opportuno usare?
La matrice di sostituzione più semplice considera solo il criterio di identità ed è pertanto costituita esclusivamente da valori 1 in corrispondenza della diagonale e 0 in tutte le altre posizioni. Oggi, per allineare sequenze proteiche vengono utilizzate quasi esclusivamente matrici basate sulla frequenza osservata di sostituzioni in famiglie di proteine omologhe:
- le matrici PAM
- le matrici BLOSUM