1. Introduzione al problema degli allineamenti di sequenze.
1.1 Similarità di sequenze e algoritmi di allineamento
L’allineamento dovrebbe portare all’appaiamento delle regioni simili condivise da due sequenze, ma per raggiungere questo scopo possono essere adottati criteri diversi. Il problema è che i concetti di similarità e di allineamento sono molto vaghi, finché non siano definiti dei criteri ben precisi per misurare la similarità.
- Non si possono allineare sequenze senza definire dei criteri di similarità
- Per valutare quanto due sequenze siano simili tra loro si devono allineare
Per allineare due sequenze è necessario:
- Definire dei criteri di similarità;
- Disporre di un metodo, che sulla base dei criteri di similarità sia in grado di produrre un allineamento, in pratica un algoritmo (metodo di calcolo descrivibile con un numero finito di regole, che conduce ad un risultato dopo un numero finito di operazioni).
1.2 Allineamenti di sequenze biologiche con gap
La complessità del problema di allineare sequenze di acidi nucleici e di proteine deriva principalmente dal fatto che deve essere considerata la possibilità che il miglior allineamento comporti l’inserimento di gap nelle sequenze.
Esempio
Senza gap
I P L M T R W D Q E Q E S D F G H K L P I Y T R E W C T R G
| | | | | | | | | |
C H K I P L M T R W D Q Q E S D F G H K L P V I Y T R E W
Con gap
I P L M T R W D Q E Q E S D F G H K L P - I Y T R E W C T R G
| | | | | | | | | | | | | | | | | | | | | | | | |
C H K I P L M T R W D Q - Q E S D F G H K L P V I Y T R E W
I due allineamenti alternativi su scritti sono stati prodotti rispettivamente senza e con la possibilità di inserire gap e appare evidente come inserendo un gap in ciascuna delle due sequenze si passi da 10 a 25 appaiamenti esatti.
L'inserimento di gap comporta principalmente due complicazioni:
- La prima relativa alla definizione di criteri di similarità (risolvibile attribuendo un'opportuna penalità ad ogni gap);
- La seconda alla possibilità di disporre di algoritmi adeguati.
Un algoritmo di scorrimento di una sequenza sull’altra è assolutamente inadatto allo scopo perché ci sono troppi modi con cui si può inserire un gap nelle sequenze da allineare.
In una sequenza lunga n caratteri è infatti possibile inserire un singolo gap in n-1 posizioni generando n sequenze diverse. Consentendo un numero peggiore di gap il numero di possibili sequenze aumenta in modo esponenziale.
Quindi è necessario pensare ad algoritmi di allineamento più efficienti.
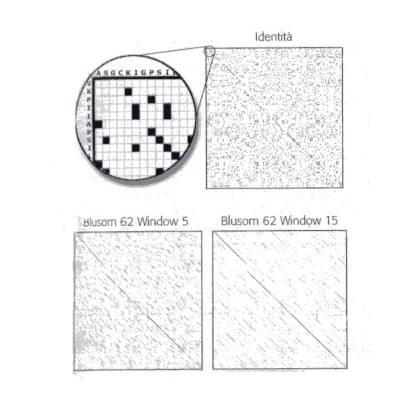
1.3 Visualizzazione di dot matrix
Le due sequenze da confrontare sono poste ai margini di una matrice di allineamento. Una sequenza è scritta da sinistra a destra in corrispondenza del margine superiore, l’altra dall’alto in basso in corrispondenza del margine sinistro. Quindi ogni casella della matrice fa riferimento a due lettere, una appartenente ad una sequenza e una all’altra. Nel caso più semplice, se le due lettere sono uguali la casella sarà colorata in nero e apparirà come un punto (dot) all’interno della matrice, altrimenti resterà bianca. In questo modo ogni allineamento di una certa lunghezza apparirà come un segmento diagonale e sarà visivamente distinguibile.
Le dot matrix sono molto convenienti per l’immediatezza e l’intuitività con cui spesso riescono a fornire indicazioni utili, tuttavia non esprimono un valore dell’allineamento, ma solo la sua rappresentazione grafica. Esse, inoltre, consentono di individuare e localizzare similarità di sequenza anche in presenza di gap che graficamente appaiono come salti in diagonale, e permettono un facile riconoscimento di regioni ripetute, che appaiono come segmenti diagonali paralleli.
2. Le matrici di sostituzione
Confrontando gli aminoacidi, è opportuno creare dei criteri di similarità che non si basino solo sull’identità assoluta, ma sulle loro caratteristiche chimico-fisiche.
Le matrici di sostituzione comprendono 210 valori: 20 (sulla diagonale) relativi al punteggio da attribuire all’appaiamento di ogni aminoacido con se stesso, gli altri 190 relativi a tutte le possibili sostituzioni aminoacidiche. Spesso i 190 valori sono riportati per comodità anche nella loro parte speculare.
La matrice di sostituzione più semplice considera solo il criterio di identità ed è pertanto costituita esclusivamente da valori 1 in corrispondenza della diagonale e 0 in tutte le altre posizioni. Una semplice matrice di sostituzione è una matrice 20x20 in cui per ogni posizione si riporta il punteggio di quella specifica coppia data un’osservazione generale.
Un meccanismo di punteggi che voglia tenere in considerazione solo le identità e non le similitudini, potrà sempre utilizzare una matrice con valori alti nella diagonale principale e 0 nelle altre.
Oggi per allineare sequenze proteiche vengono utilizzate quasi esclusivamente matrici basate sulla frequenza osservata di sostituzioni in famiglie di proteine omologhe:
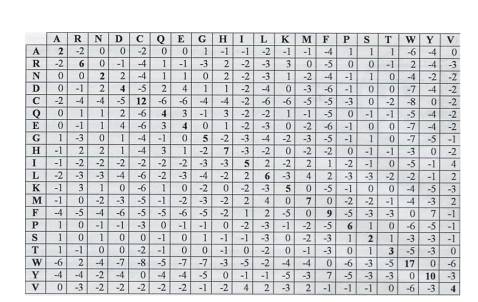
La figura rappresenta la matrice di sostituzione PAM240.
Questa matrice è spesso utilizzata per la ricerca di similarità in sequenze filogeneticamente distanti.
3. Metodi di allineamento esatto
La scelta dell’algoritmo dipende anche dalle finalità che l’allineamento si propone:
- allineamento globale
- allineamento locale
Dal punto di vista strettamente computazionale, ovviamente, l’allineamento migliore è quello che totalizza il punteggio maggiore.
Da un punto di vista puramente biologico i criteri di similarità privilegiano gli allineamenti locali.
L'allineamento esatto prevede due metodi:
- Utilizzo di algoritmi basati sulla
programmazione dinamica implementati:
- nel metodo di Needleman e Wunsch per la ricerca dell'allineamento globale
- nel metodo di Waterman e Smith per la ricerca dell'allineamento locale
- Utilizzo di algoritmi euristici (approssimati)implementati nei metodi di Database Similarity Searching (FASTA e BLAST)
4. Metodi euristici di allineamento
Database similarity searching:
Confronto di una sequenza a funzione incognita con sequenze a funzione nota depositate nelle banche dati biologiche.
Prerequisiti dei metodi di Database similarity searching
- Velocità
- Sensibilità
- Selettività
Fissata una soglia minima di similarità se
TP = positivi veri
PP = positivi predetti (falsi e veri)
AP = actual positives (n. reale di sequenze omologhe presenti nel db indipendentemente dalla soglia)
Sensibilità = TP/AP
Selettività = TP/PP
La scelta della soglia è fatta su basi statistiche ed è determinante.
Alta sensibilità bassa selettività e viceversa.